Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
In the early stage of the C. elegans sequencing project, the ab initio gene prediction program Genefinder was used to find protein-coding genes. Subsequently, protein-coding genes structures have been actively curated by WormBase using evidence from all available data sources. Most coding loci were identified by the Genefinder program, but the process of gene curation results in a continual refinement of the details of gene structure, involving the correction and confirmation of intron splice sites, the addition of alternate splicing forms, the merging and splitting of incorrect predictions, and the creation and extension of 5’ and 3’ ends. The development of new technologies results in the availability of further data sources, and these are incorporated into the evidence used to support the curated structures. Non-coding genes are more difficult to curate using this methodology, and so the structures for most of these have been imported from the literature or from specialist databases of ncRNA data. This article describes the structure and curation of transcribed regions of genes.
Sydney Brenner, the founder of modern worm biology, once said, “Old geneticists knew what they were talking about when they used the term ‘gene’, but it seems to have become corrupted by modern genomics to mean any piece of expressed sequence…” (Brenner, 2000). Dr. Brenner's lament serves to illustrate two points: the first is that the concept of a gene can mean different things to different people in different contexts, the second is that the concept of a gene has been evolving, not only in the modern genomic era, but ever since it first appeared in the early 1900s as a term to conceptualize the particulate basis of heritable physical traits (Snyder and Gerstein, 2003). Therefore, in a review of gene structure in C. elegans it seems prudent to define what we mean by a gene.
Our definition of a gene is essentially: “a union of genomic sequences encoding a coherent set of potentially overlapping functional products” (Gerstein et al., 2007). This encompasses promoters and control regions necessary for the transcription, processing and if applicable, translation of a gene. Hence, we include not only protein-coding genes (genes that encode polypeptides), but also non-coding RNA genes (ribosomal RNA, transfer RNA, micro RNA, anti-sense RNA, piwi-interacting RNA, and small nuclear RNA genes). One additional type of gene we will briefly discuss is the pseudogene, though these are not usually considered to be functional.
The full extent of most C. elegans genes is not known because promoters remain, for the most part, incompletely defined. Even the full extent of the primary transcript is frequently not known because a majority (70%) of protein-coding genes are rapidly modified by trans-splicing, which involves the addition of a short 22 nt exogenous RNA species to the 5’ end of a transcript (Zorio et al., 1994). Definition of the true 5’ ends of genes is an active area of research (Chen et al. 2013; Kruesi et al. 2013; Saito et al. 2013; Gu et al. 2012). Some non-coding RNA genes are also trans-spliced; a precursor of the microRNA let-7 (C05G5.6) was identified with a trans-splice leader sequence (Bracht et al., 2004). This article is concerned primarily with the properties of the transcribed regions of C. elegans genes.
In the initial stage of the C. elegans sequencing project, prior to the publication of the genome in 1998 (The C. elegans Sequencing Consortium, 1998), Genefinder (Green and Hillier, unpublished software) was the gene prediction program of choice. Genefinder is an ab initio predictor and requires only a genomic DNA sequence and parameters based on a training set of confirmed coding sequences. Note that Genefinder, like most other gene prediction tools, is actually a coding sequence (CDS) predictor and does not attempt to define untranslated regions (UTRs).
In the WormBase database, the structure of a coding gene is held as three different types of data. The first is the “Gene”, which holds information on the span from the start to end of the transcribed region of that locus. The second is the “Transcript”, which holds the exon structure, including the 5’ and 3’ untranslated regions (UTRs) and attempts to faithfully model a mature mRNA sequence. The third is the “CDS”, which is purely a protein-coding set of exons from a START codon to a STOP codon, with no UTR. It is only the “CDS” structure which is manually curated, with often two or more “CDS” isoforms being made for the same gene, based on evidence for trans-splicing or alternative splicing. The “Transcript” structures are automatically deduced by combining the “CDS” structures and the aligned EST, mRNA and OST transcript data to extend the “CDS” out to cover the UTRs. The predicted “Transcript” structures for each locus are then combined to find the maximum bounds of transcription for that locus, giving the reported span of the “Gene”. Thus the manually curated “CDS” structures are used to automatically define the “Transcript” structures and “Gene” regions. RNAseq data is not currently used to extend the UTR regions of the “Transcript” structures because their short length often makes it hard to distinguish from which of two nearby genes they were derived.
With the completion of the C. elegans genome sequence, a renewed effort to improve the coding gene predictions was initiated. The coding sequence (CDS) structures are manually curated, using all available evidence from the literature, protein similarity, peptides identified by mass-spectrometry, transcript evidence, ab initio gene predictions and features such as trans-spliced leader sequence (TSL) sites and polyadenylation sites.
Even for a model organism with a mature reference genome sequence such as C. elegans, the definition of a canonical set of gene structures is constantly being revisited. As new projects and technologies producing transcript and other data have appeared, WormBase has used them to improve the CDS structures. Some of these include:
• RNASeq short read data, extracted from the NCBI and EBI Short Read Archive. These data are from many projects with about half coming from the various releases of data from the modENCODE project (Gerstein et al. 2010).
• messenger RNA (mRNA) deposited with the International Nucleotide Sequence Database Collaboration (INSDC).
• Expressed Sequence Tag (EST) from both C. elegans (Yuji Kohara, unpublished; http://nematode.lab.nig.ac.jp/) and other nematode species (from multiple sources including INSDC, the Genome Institute of Washington University St. Louis, NemBase, available at www.nematodes.org and Nematode.net).
• Orfeome Sequence Tags (OST) (Reboul et al., 2003; Vaglio et al., 2003).
• Race Sequence Tags (RST) (Marc Vidal laboratory 2009, unpublished).
• cDNA data from other projects (e.g., Makedonka Mitreva 2008, unpublished).
• mass spectroscopy projects, primarily from the Michael MacCoss and Michael Hengartner laboratories.
• projects looking for trans-spliced leader sequences sites (TSLs) (Lamm et al. 2011; Hwang et al. 2004).
• polyadenylation sites from EST sequences and several projects (Jan et al. 2011; Lamm et al. 2011; Mangone et al. 2010).
• the use of transcription start sites is an active area of research (Chen et al. 2013; Kruesi et al. 2013; Saito et al. 2013; Gu et al. 2012).
The RNASeq, EST, mRNA, OST, RST and other transcript data are aligned to the genome and are used to confirm the splice donor and acceptor sites of introns and guide the curation of alternatively spliced CDS isoforms, although the sparsity of long transcript evidence in some genes with low levels of expression can make it difficult to confirm some putative isoform structures.
The various features that indicate the start and end of a gene, including regulatory regions, TSS sites, TSL sites, expressed transcripts that define the UTRs, polyadenylation sites, and homologous gene structures are all valuable evidence when curating the structure of a gene.
To direct curation efforts towards the genes with the lowest-quality gene models, a system of automatically comparing the structure of the CDS against the available alignment evidence is used. Those structures having the greatest discordance with the alignment evidence are assigned a higher priority for manual curation (Williams et al. 2011).
Through manual curation, we endeavor to continually improve the accuracy of the gene structures. How accurate are the gene structures? In WormBase release WS237 (May, 2013), 12,530 CDS structures (48%) have every base of every exon confirmed by either mRNA, EST, or OST transcription evidence, showing the gene is real and the structure correct. An additional 11,602 (44%) have at least one base of an exon confirmed by transcript data, indicating the gene is real and part of the structure is correct. The remaining 8% of the CDS predictions currently have no mRNA, EST or OST evidence and tend to be expressed at a low level and so lack good RNASeq evidence. These gene structures are therefore largely based on ab initio gene prediction or sequence conservation with orthologs or paralogs such as the family of genes coding for seven-transmembrane domain receptors. The above statistics do not currently use coverage by RNASeq short reads to confirm that the full structure is correct. The short length of RNASeq reads can give rise to unreliable alignments which warrants their treatment as a separate class of data from the ESTs/mRNAs (which are longer and higher quality). As technology underlying RNASeq data improves, resulting in longer read lengths and higher quality, this distinction will become less necessary.
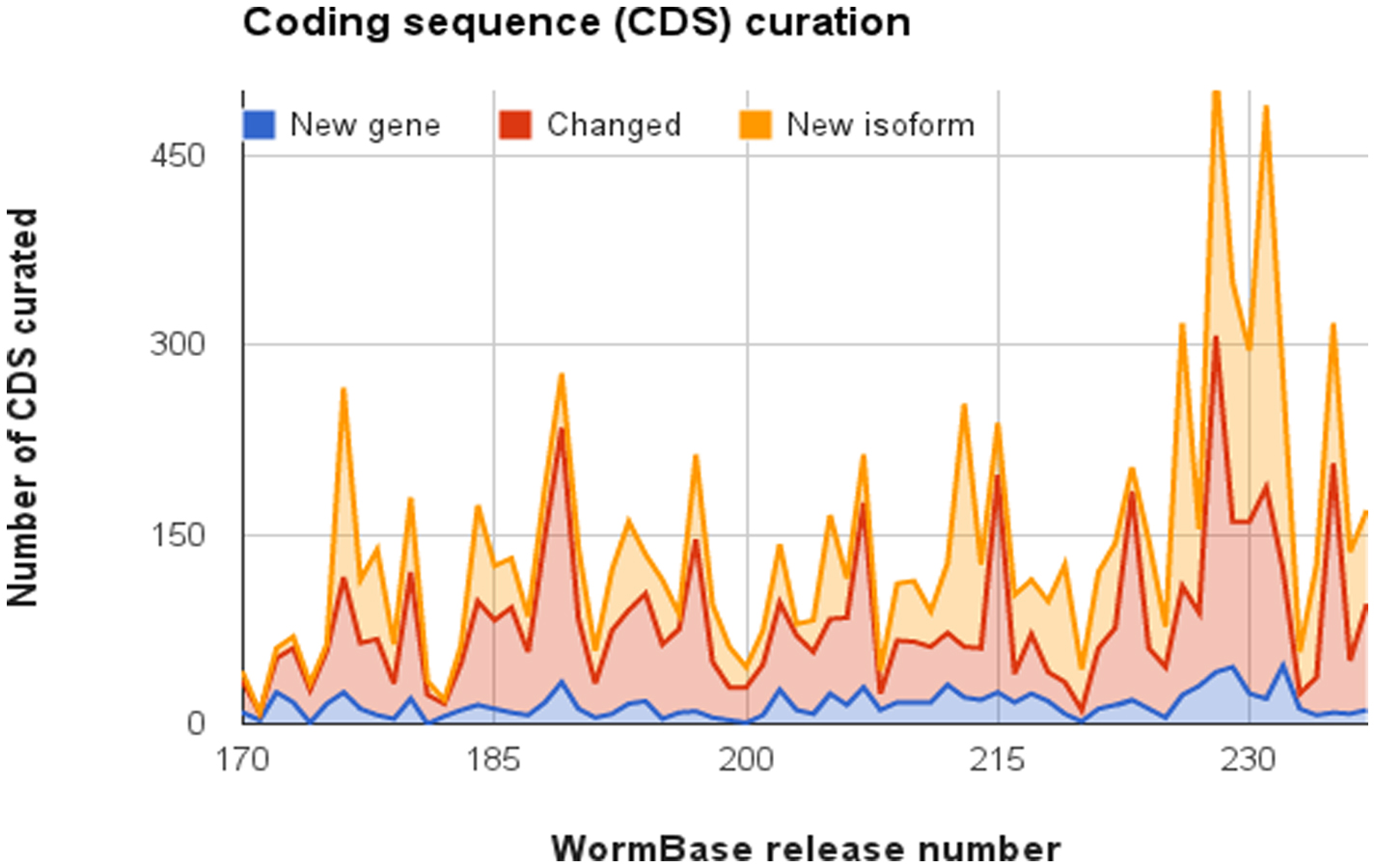
How many additional C. elegans genes remain to be found? It's hard to know for certain, but over the 30 months from release WS220 to release WS237, the number of curated protein-coding genes increased by 119, some of which were the result of splitting existing genes. In release WS237, all of the new genes that could be unambiguously supported by the evidence from the modENCODE RNASeq data had been curated leaving about 140 which are more dubious. It is expected that as experimental evidence accumulates, the curated CDS structures will be refined and the number of new genes and structural changes that can be easily made will decline to low levels, with changes mainly based on detailed investigation of the properties of a gene in the literature. At present, however, the number of changes to existing CDS structures, creation of new coding genes and creation of new isoforms of existing genes made in each WormBase release does not appear to be decreasing (Figure 1). This is largely due to the continuing incorporation of the final 2012 freeze of the modENCODE RNASeq data. The modENCODE gene models are a useful guide when curating CDS structures but have not been converted into curated CDS structures en masse. This is because many curated WormBase CDS structures incorporate knowledge from experts or the literature and should not be overwritten automatically and also the modENCODE gene model structures were based, in part, on a specific older set of WormBase gene structures, many of which would have been incorrect before the advent of large-scale RNASeq data. Figure 2 shows some typical changes to genes structures involving merging and splitting of genes.
|
Figure 1. Manual curation of gene structures. This area graph shows the creation of new coding genes, changes made to CDS structures, and creation of new coding isoforms in each release from WS170 (June, 2006) to WS237 (May, 2013).
|
Figure 2. Examples of curational changes made to gene structures. (A) Two genes are merged to make one gene and the final intron has the splice 3’ site changed. (B) One gene is split into two genes. (C) Two genes are restructured to move some exons from the second to the first. The second gene (Y19D10B.8) was previously curated in release WS88 (in 2002) before more recent evidence was available.
There are currently 321 protein-coding gene models that have been classified as belonging to a transposable element. These Transposon-CDSs have been classified as such based on published associations, protein similarity to transposon protein domains, or overlap between these loci and repeat motifs corresponding to transposable elements. An example Transposon-CDS is B0213.1. This gene belongs to a member of the Tc1 transposon family which was the first C. elegans transposable element identified (Rosenzweig et al. 1983). WormBase excludes Transposon-CDSs from the canonical protein-coding gene set, so they are not treated as a part of the normal set of coding genes and they do not have their protein translation added to the set of available WormBase proteins.
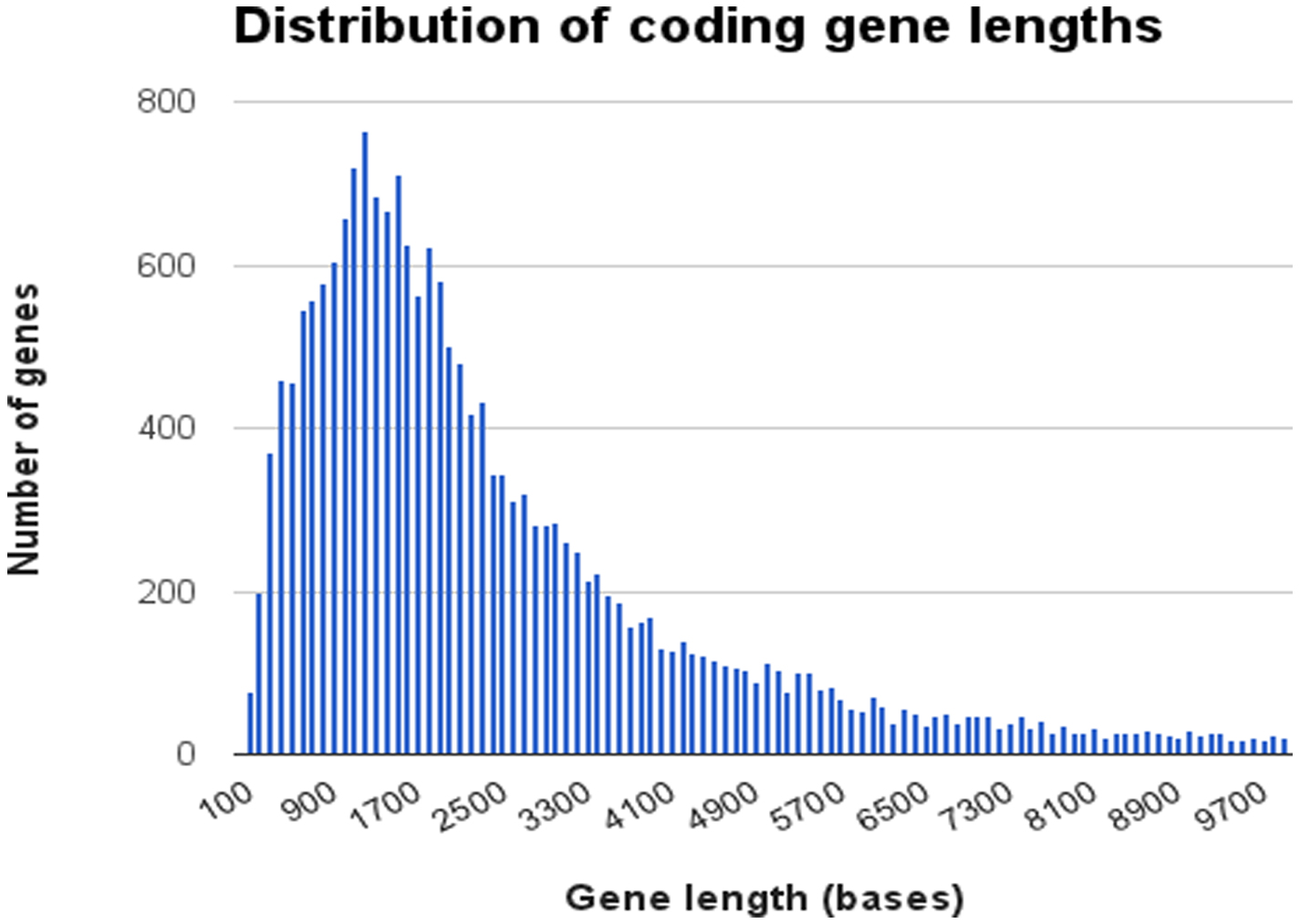
In WormBase data release WS237, there were 20,512 protein-coding genes. Most coding genes are relatively small (Figure 3), covering a genomic region of approximately 3 kbp (from start to stop codon including introns), although there are some very large genes which skew the mean. The median size of coding genes is only 1,956 bp, spanning from K07C5.13 (81 bp) and F11A6.15 (87 bp) to a few genes with very large spans caused by introns of more than 100 Kb such as Y61B8A.6 (134,155 bp) and nhr-27 (F16H9.2) (102,626 bp) and the C. elegans titin gene ttn-1 (W06H8.8) with a span of 81,718 bp. Figure 4 shows the distribution of sizes of the coding sequence (CDS) part of coding genes which hints at being a bimodal curve.
|
Figure 3. Distribution of coding gene lengths. Data was obtained from WormBase release WS237. Each data point represents the number of genes in each size class in increments of 100 bp
|
Figure 4. Distribution of CDS sizes. This is the lengths of the CDS coding regions of genes from the START codon to the STOP codon (inclusive) without the introns. Data was obtained from WormBase release WS237. Each data point represents the number of CDSs in each size class in increments of 100 bp. Where there are several CDS isoforms in a gene, they are all counted.
C. elegans genes, like most eukaryotic protein-coding genes, contain exons separated by introns. There are 131,083 unique, coding exons (the same exon used in alternatively-spliced isoforms of the same gene is considered as one unique exon) in the WS237 protein-coding gene set, which account for 26% of the genome, considerably more than the corresponding estimate of 1.5% for the human genome (The International Human Genome Sequencing Consortium, 2001).
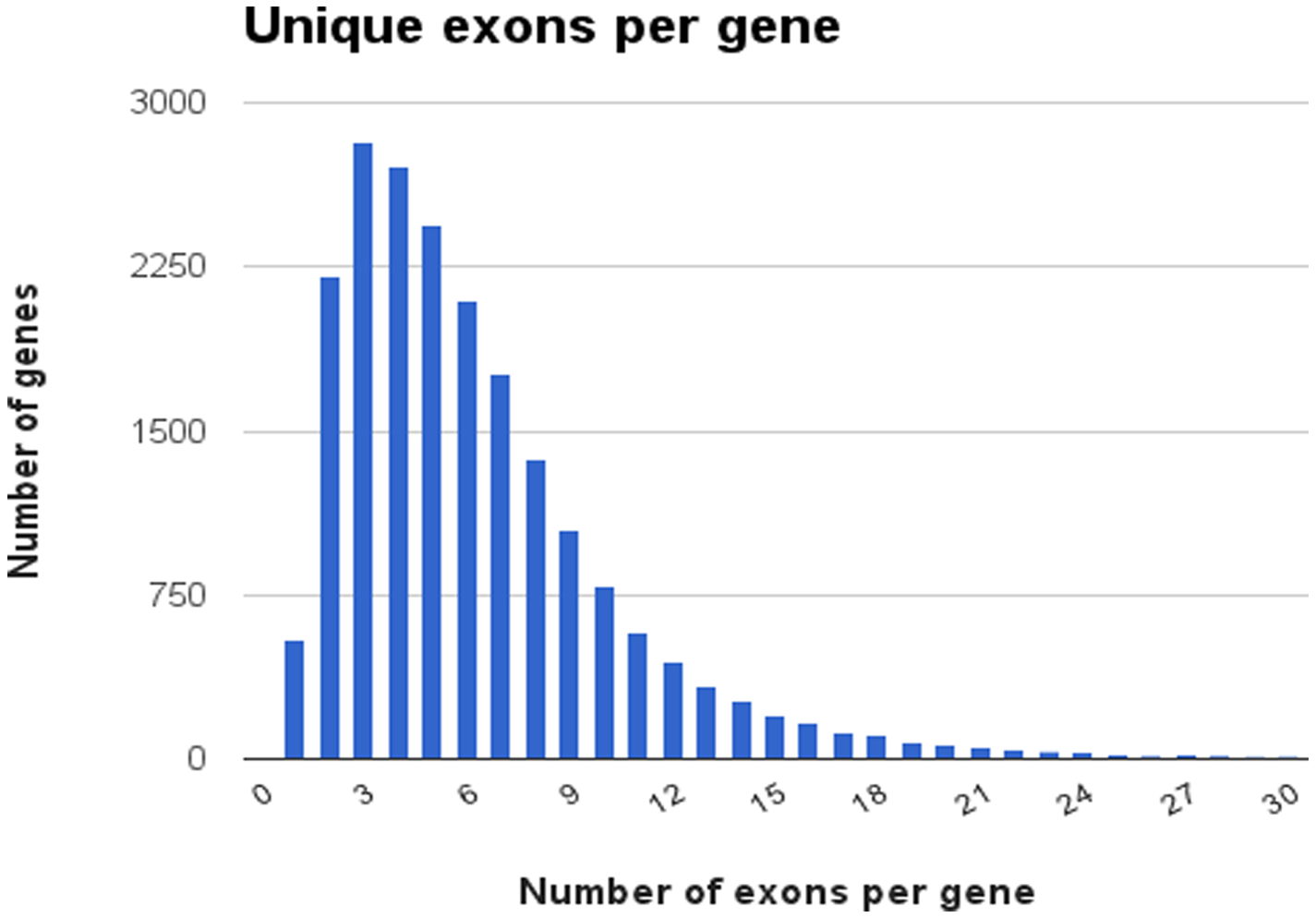
The average gene contains 6.4 coding exons, although there are a few genes with a large number of exons (Figure 5). W06H8.8g, an isoform of the titin gene (ttn-1), has 66 coding exons and there are 73 unique exons used overall in the various titin isoforms.
|
Figure 5. Distribution of the number of exons per gene. Data was obtained from WormBase data release WS237. Each bar represents the number of genes having a specific number of unique exons.
The average size of unique exons in all protein-coding genes is 200.7 bp, but there are a small number of very large exons. Again, as with gene size, these few large exons skew the average. The median size is only 123 bp, thus exons are similar in size to exons in human genes (The International Human Genome Sequencing Consortium, 2001). An example of a large exon is one of 7,569 bp found in pqn-43 (F54E2.3b). Some examples of small exons include a 7 bp exon in dyf-6 (F46F6.4d) and a 9 bp exon in grld-1 (F29C4.7c).
There are 549 single-exon genes in WS237 that comprise 2.7% of all coding genes and 0.87% of the total coding bases in all coding genes. The curation of single-exon genes is particularly prone to error, because they may be a part of a non-coding gene or a 3’ UTR that has evidence of expression with an open reading frame that can be marked up as a coding region.
There are 108,151 (WS237) unique introns (the same intron used in different isoforms or spliced variants is considered as one unique intron) in all of the coding sequences of C. elegans. Of these, 100,065 (92.5%) are confirmed by RNASeq with 3 or more reads in a library spanning the intron boundaries, and 88,174 (81.5%) are confirmed by EST or cDNA sequences spanning them.
• 85,658 (79.2%) are confirmed by both RNAseq and EST/cDNA.
• 14,407 (13.3%) have only RNASeq confirmation.
• 2,516 (2.3%) have only EST/cDNA confirmation.
• 5,570 (5.1%) have no direct EST/cDNA or RNASeq transcript evidence.
The introns with no supporting evidence or evidence from only RNAseq or only EST/cDNA data are usually from poorly expressed genes which consequently have sporadic direct evidence based on expression. Those with no direct evidence are based on either ab initio predicted structures or have other supporting evidence such as homology to other genes.
Work is continuing to curate novel alternately spliced introns that have been confirmed by RNAseq, mass-spectroscopy and other projects.
The most common size of CDS introns is 47 bp with the median size being 65 bp. There are some examples of very small artificial introns in WormBase used by the gene sequence curators to change the frame of a CDS. In H37A05.4 this is used to work around a strongly suspected, but unverified, genomic sequence error in a highly repetitive region. In ZK484.1a there is a conserved, specific +1 translational frameshift to express the orthinine decarboxylase antizyme protein. In lys-9 (C54C8.6) there is suspected to be post-transcriptional modification to correct the translation frame. There are three natural small introns with a size of 25 bp or less: xbp-1 (R74.3a) (23 bp), K08E5.6 (25 bp), and E04A4.3 (25 bp).
The largest confirmed intron is 100,912 bp, found in nhr-27 (F16H9.2b), supported by an RST sequence indicating that there is an SL1 trans-spliced isoform which re-uses 34 bp of coding sequence contained within gei-3 (T22H6.6a). The next longest intron is 21,230 bp, found in kin-1 (ZK909.2j). It is confirmed by a cDNA and an EST clone containing the SL1 trans-splice leader sequence at its 5’ end.
The intron size in C. elegans appears to be positively correlated with local recombination rates (Prachumwat et al., 2004) and short introns are preferentially found in highly expressed genes (Castillo-Davis et al., 2002).
The introns of C. elegans have always been considered small, but as more genomes are being sequenced and annotated it is becoming evident that they are not distinctly smaller than those of most eukaryotes. The modal length of Drosophila introns is only 59 bp (The International Human Genome Sequencing Consortium, 2001), as compared to 47 bp for the worm. The mean size of introns on the largest, somatic, macronuclear chromosome of Paramecium is only 25 bp (Zagulski et al., 2004). Fungal introns are also small; Neurospora introns average 134 bp (Galagan et al., 2003), Sordaria macrospora 106 bp (Nowrousian et al., 2004), and Cryptococcus neoformans 67 bp (Loftus et al., 2005). Even in humans the modal intron length is only 87 bp, but there are also some very large introns, shifting the mean length to more than 3,300 bp genes (The International Human Genome Sequencing Consortium, 2001).
C. elegans introns follow the GU-AG splice site rule, although GC is a rare 5’ splice site variant (Blumenthal and Steward, 1997). There are 870 introns with GC-AG splice sites in WS237. A well-characterised example is the donor splice site of intron 8 of let-2 (F01G12.5b) (Sibley et al. 1993). From their analysis of 669 introns Blumenthal and Steward found that C. elegans has a highly conserved and extended 3’ splice site (UUUCAG) and no obvious polypyrimidine tract other than the 3’ splice site consensus. They suggest that the 3’ intron boundary may be more important in C. elegans intron recognition than in other organisms.
In addition to splicing information, some C. elegans introns contain sequences involved in the regulation of gene expression (Zhang and Emmons, 2000).
Alternative splicing is covered in detail in the WormBook chapter Pre-mRNA splicing and its regulation in Caenorhabditis elegans, so here we will just mention it briefly. Alternative splice forms are only annotated in WormBase when there is direct transcript or literature citation evidence for the alternative form. In WormBase release WS237 there were 20,512 coding gene loci, 3,623 (18%) genes had curated evidence of alternative splicing with a total of 9,484 alternatively-spliced forms giving rise to different protein products (Figure 6). The number of alternatively-spliced forms per gene tends to be small with 68% having either one or two alternatively-spliced forms. The remaining 32% of loci have between 3 and 17 alternatively-spliced forms. Many of these alternative forms show only minor changes to the CDS with alternate 5’ end structures (SL1/2 outrons) or small multiples of 3 (i.e., 3,6,9) bp differences to the splice donor or acceptor sites.
Processed pseudogenes, which are created by reverse transcription of mRNA into cDNA followed by reintegration into the genome, are fairly easy to detect because they lack introns. These are rare in C. elegans (Harrison et al., 2001). Unprocessed pseudogenes arise by duplication of a gene, which is subsequently disabled by random mutation. Unprocessed pseudogenes usually have features that aid in their identification, such as frameshifts, premature stops, insertions and truncations compared to their functional homolog, or a ratio of non-synonymous vs. synonymous nucleotide substitution rates indicating a lack of purifying selection. These features are probably valid indicators for most pseudogenes, assuming that the function of the gene is at the protein level. Some pseudogenes are expressed, but mRNAs containing premature stops are usually subject to rapid, nonsense-mediated decay (NMD), so they were difficult to detect using traditional sequencing technologies. With the advent of new sequencing technologies it is possible to detect transcripts that are unique to a pseudogenic loci compared to their functional homolog.
The gene ZK637.6 (Park et al 2013) is an example of how regarding pseudogenes as simply inert, broken genes should be treated with caution. This is a pseudogene with a frameshift and a premature STOP codon when compared to the first exon of the parental gene, asna-1 (ZK637.5), a positive regulator of insulin secretion. The miRNA transcript mir-249 (Y48D7A.3) binds to the ZK637.6 transcript and directs its cleavage. There is a strong negative correlation between expression of the ZK637.6 transcript and mir-249. The complementarity of mir-249 and ZK637.6 is evolutionarily conserved in nematodes, which may indicate a possible function of the miRNA in degrading the pseudogene's transcript. There is currently no evidence, however, of a functional role for the pseudogene. The expression of the asna-1 mRNA is not dependent on the expression of mir-249, so the pseudogene is not acting as a “miRNA decoy”, guarding its parental gene by acting as the regulated target.
Even before large-scale sequencing of the C. elegans genome commenced, pseudogenes were identified in the major sperm protein (MSP) (Ward et al., 1988) and heat-shock protein gene families (Heschl and Baillie, 1989). Genefinder predictions associated with the genome sequencing project did not attempt to predict pseudogenes. The first genome-wide analysis of pseudogenes was done in 2001 (Harrison et al., 2001). Analyzing Wormpep release 18 and the corresponding version of the genomic sequence (which had only 332 annotated pseudogenes), the authors found 2,168 pseudogenes, or 11.7% of the annotated genes. Most of these were unprocessed pseudogenes, with only 208 designated as processed pseudogenes. They found that pseudogenes are unevenly distributed across the genome with a disproportionate number on chromosome IV; the density was also higher on chromosome arms than in the central regions. Looking at the distribution of pseudogenes among gene families they found that the number of pseudogenes is not correlated with the size of the gene family, but several families were associated with large numbers of pseudogenes. One of these families was the 7-TM receptor family, a finding supported by Robertson's characterization of chemoreceptor gene families (Robertson, 2002; Robertson, 2000; Robertson, 1998). A higher estimate of the number of pseudogenes comes from an analysis of reporter gene fusions (Mounsey et al., 2002). Extrapolating from the number of 364 randomly selected reporter gene fusions that showed no expression, the authors estimate that 20% of the annotated C. elegans genes may be pseudogenes. Furthermore, they found that pseudogenes were enriched for genes that had been recently duplicated.
WormBase release WS237 contains 1469 annotated pseudogenes (these are not counted as part of the set of 20,512 protein coding genes). This is lower than either of the above estimates, however coverage is closer to these estimates thanks partly to WormBase staff being able to work with modENCODE data providers (Gerstein et al. 2010). Almost half of the curated loci are located on chromosome V (Table 1). This bias is partly a reflection of the curation of chemoreceptor genes, which are located primarily on chromosome V (Robertson, 2002; Robertson, 2000; Robertson, 1998; see also The putative chemoreceptor families of C. elegans). There have been numerous attempts at automating the identification of new and mis-classified pseudogenes in C. elegans, and the number of annotated pseudogenes in WormBase is still likely to be an underestimate.
Table 1. Chromosomal distributions of protein-coding genes, ncRNAs, and pseudogenes. Gene data was taken from WormBase release WS237. Chromosomal cluster boundaries were taken from Barnes et al. (1995).
| Chromosome | Size (Mb) | Protein-coding genes | ncRNA genes | Pseudogenes | Density (genes/Mb) |
|---|---|---|---|---|---|
| I | 15.08 | 2,920 | 1,117 | 143 | 277 |
| Left | 4.00 | 600 | 201 | 9 | 202 |
| Center | 6.26 | 1,381 | 551 | 27 | 312 |
| Right | 4.82 | 939 | 365 | 107 | 292 |
| II | 15.28 | 3,552 | 1,403 | 186 | 336 |
| Left | 5.90 | 1,555 | 429 | 90 | 351 |
| Center | 5.44 | 1,277 | 669 | 42 | 365 |
| Right | 3.94 | 720 | 305 | 54 | 273 |
| III | 13.78 | 2,699 | 1,019 | 106 | 277 |
| Left | 4.80 | 843 | 325 | 59 | 255 |
| Center | 4.29 | 1,009 | 374 | 8 | 324 |
| Right | 4.68 | 847 | 320 | 39 | 257 |
| IV | 17.49 | 3,348 | 15,990 | 276 | 1,121 |
| Left | 6.74 | 1,231 | 3,212 | 64 | 668 |
| Center | 5.08 | 1,207 | 848 | 66 | 417 |
| Right | 5.67 | 910 | 11,930 | 146 | 2,290 |
| V | 20.92 | 5,149 | 1,971 | 687 | 373 |
| Left | 6.51 | 1,533 | 407 | 90 | 311 |
| Center | 6.99 | 1,798 | 1,059 | 106 | 423 |
| Right | 7.42 | 1,818 | 505 | 491 | 379 |
| X | 17.72 | 2,844 | 3,088 | 86 | 338 |
Non-coding RNAs are classified into 11 identifiable types. 61% of non-coding loci (15,366) were identified through large scale deep sequencing and correspond to the piRNA type (Ruby et al. 2006). These are primarily clustered on chromosomes IV and V which accounts for the bias on Table 1. The remaining non-coding RNAs come from a variety of sources: expert databases, publications, and WormBase curators.
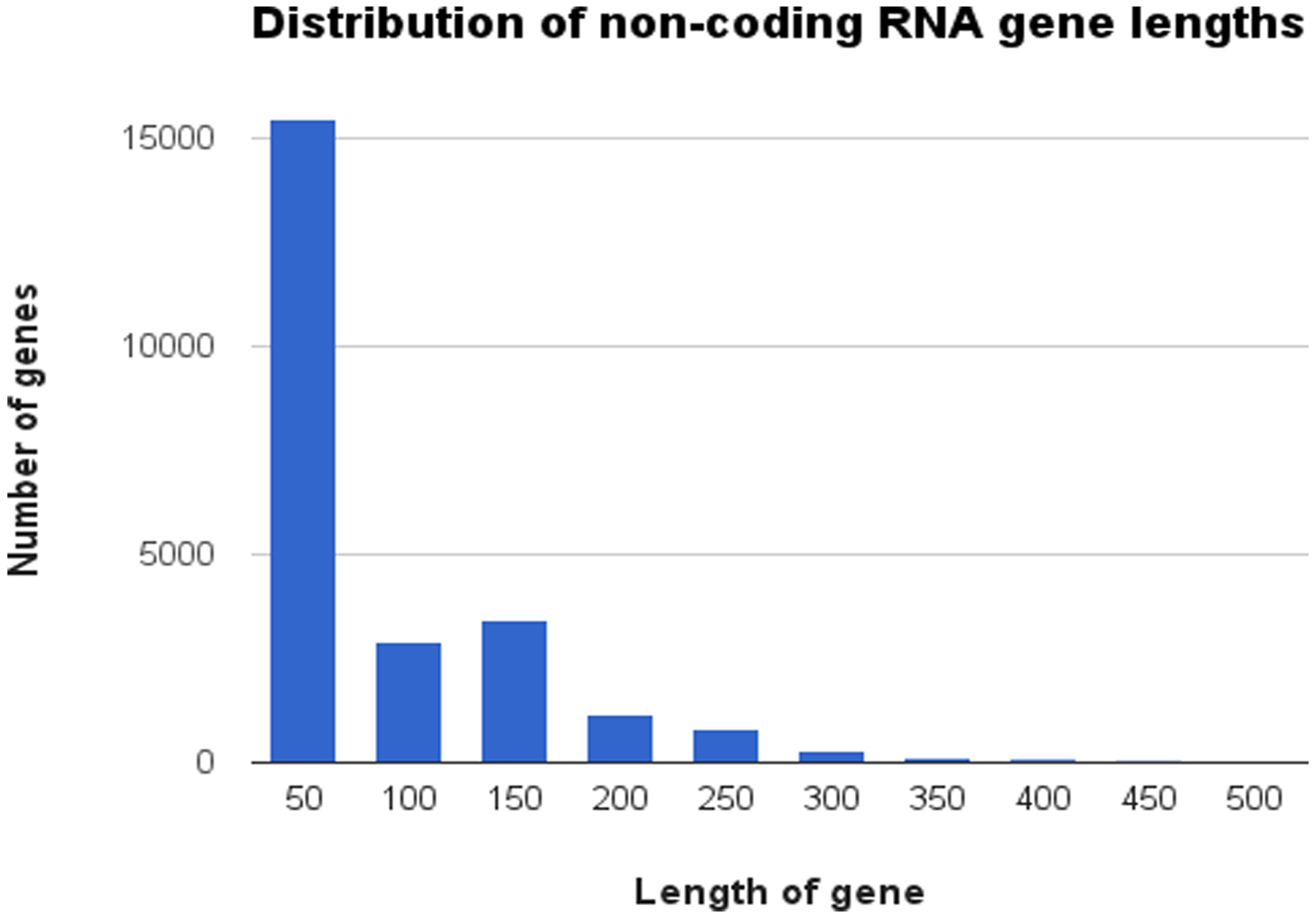
In WS237 there were 24,611 annotated ncRNA loci. Most (97%) of the C. elegans non-coding RNA genes are less than 300 bp long and they have a range from 17 bp (three members of a class of tiny non-coding RNA (tncRNA) genes: Y37E3.24, W04A8.8, and F02C12.7) to lincRNAs such as Y105C5B.1420 and linc-136 (T27C4.6) with genomic spans of 8,360 bp and 6,035 bp respectively (Figure 7). There is an unusual number of ncRNA gene in the size range 100 to 150 bp. 3030 of the 3426 genes in this size range are from a modENCODE project which aimed to identify ncRNA genes using RNAseq (Gerstein et al. 2010). It is possible that this experiment particularly targeted ncRNA genes of the order of the size of a RNASeq short read, resulting in a bias towards the identification of genes of this size.
|
Figure 7. Distribution of non-coding RNA gene lengths. Data was obtained from WormBase data release WS237. Each bar represents the number of genes in each size class in increments of 50 bp
In 2012 a study was conducted to investigate the existence of long intergenic non-coding RNA genes (lincRNA) outside of vertebrates (Nam, et al. 2012). This study took all available RNASeq, polyadenylation site, and ribosome-mapping data to identify lincRNA-like structures in C. elegans. Hundreds of candidate lincRNAs were proposed and then were scrutinised by WormBase, resulting in 170 structures. Approximately 25% of the newly identified lincRNAs showed little sequence conservation and mapped antisense to clusters of 22G or 26G endogenous siRNAs, as would be expected if they serve as templates and targets for these siRNAs. The other 75% displayed greater conservation and included lincRNAs with expression and sequence features associating them with processes such as dauer formation, male identity, sperm formation, and interaction with sperm-specific mRNAs (Nam, et al. 2012).
The antisense RNA genes (asRNA) form a relatively new class of non-coding RNA genes annotated in WormBase and forms a sub class within the lincRNAs. This class currently only consists of the published gene set (Nam, et al. 2012) named anr-1 (T26A5.11) to anr-58 (Y50D4A.9) that were discovered by utilising available RNASeq, polyadenylation site, and ribosome-mapping data to identify lincRNAs, with those complementary to protein-coding transcripts being classified as asRNAs.
Members of the microRNA (miRNA) class of 22-nucleotide RNAs regulate the expression of target genes containing sequences that have antisense complementarity to the mature miRNA transcript. miRNAs annotations in WormBase are almost exclusively derived from miRBase (Kozomara et al. 2011). A small number of annotations have been received via private submission but these have ultimately been incorporated into miRBase and the additional evidence incorporated into the locus annotation. In WS237 there were 227 annotated miRNA gene loci with 444 annotated transcripts. These transcripts correspond to 230 miRNA primary transcripts and 214 mature transcripts. An example of a gene that produces 2 mature transcripts is mir-1019 (M04C9.8a and M04C9.8b). This RNA appears to be the complete intronic sequence of the dyf-5 (M04C9.5) protein coding gene, which produces a hairpin structure that results in a miR-1019-5p and miR-1019-3p active product after cleavage (Ruby et al. 2007). An example of a gene that has multiple primary transcripts but only a single identifiable mature transcript is let-7, which has at least 3 primary transcripts, C05G5.6.1, C05G5.6.2, and C05G5.6.3, with C05G5.6.3 undergoing trans-splicing to SL1 as well as polyadenylation (Bracht et al 2004).
Members of the ‘non-coding RNA’ (ncRNA) class in WormBase are RNA genes whose properties or functions have not yet been identified. These are created when a region of transcription overlaps with a region of the genome where no discernible coding intron/exon structure can be annotated, or which derive from publications where the author has not described the function.
Piwi-interacting RNA genes (piRNA) are the most abundant class of small non-coding RNA in the C. elegans genome and range in size from 26 to 31 nucleotides. These have been associated with both epigenetic and post-transcriptional gene silencing of retrotransposons and other genetic elements. In WS237 there were 15,366 piRNA genes which corresponds to 61% of the non-coding gene loci. This data was incorporated from a single publication (Batista et al. 2008).
The genes for the 18S (rrn-1.1 (F31C3.7)), 5.8S (rrn-2.1 (F31C3.11)), and 26S (rrn-3.1 (F31C3.9) to rrn-3.56 (F31C3.10)) ribosomal RNAs (rRNA), first sequenced and characterized by Ellis et al. (1986), are found in a large tandem-repeat of ~55 copies on the right-end of chromosome I. Each repeat contains one copy each of the 18S, 5.8S, and 26S genes. For the purposes of the reference genome assembly, only 1 full copy of the tandem repeat was assembled and finished. The 5S ribosomal RNA genes are found in a tandem-repeat of an estimated 110 copies on chromosome V (Sulston and Brenner, 1974; Nelson and Honda, 1985). Each copy of the 5S gene (Y102A5D.10) is interspersed with copies of one of the SL1 splice-leader genes, sls-1.4 (Y102A5D.16). The current reference assembly was produced by sequencing and finishing the clones that border the cluster to the point where reads could no longer be anchored in the correct position and an overlap was found. This resulted in a compression of the repeat cluster containing approximately 15 copies of this tandem repeat. As the reference genome only contains representative tandem repeats for each cluster, the reference gene set only contains a fraction of the full set of rRNA genes (Table 2).
Table 2. Numbers of types of non-coding RNA genes in WormBase release WS237.
| Type | Number |
|---|---|
| tslRNA | 128 |
| tRNA | 634 |
| asRNA | 26 |
| miRNA | 227 |
| ncRNA (uncategorised) | 7,821 |
| piRNA | 15,366 |
| rRNA | 22 |
| scRNA | 1 |
| snoRNA | 341 |
| snRNA | 117 |
| lincRNA | 67 |
There is one small cytoplasmic RNA gene (scRNA) in C. elegans. In virtually all vertebrate cells, Ro ribonucleoproteins (RNPs) consist of the 60-kDa Ro autoantigen bound to one of several small cytoplasmic RNA molecules known as Y RNAs. Caenorhabditis elegans embryos contain only one major species of Ro RNP complex. The gene yrn-1 (F55G1.16) encodes a noncoding RNA gene with structural similarity to the vertebrate Y RNAs. yrn-1 is bound by the C. elegans Ro protein, ROP-1. The nematode RNP complex is formed by the assembly of ROP-1 and yrn-1. RNA interference of yrn-1 does not produce any detectable phenotype (Van Horn 1995).
Small nucleolar RNA genes (snoRNA) are a sub-class of snRNAs. They are small RNA molecules that play a role in RNA biogenesis and guide chemical modifications of ribosomal RNAs (rRNAs) and other tRNA genes. There are 341 identified snoRNA genes in C. elegans.
Small nuclear ribonucleic acid RNA genes (snRNA), also commonly referred to as U-RNA, are found within the nucleus. snRNA are always associated with a set of specific proteins, in complexes called small nuclear ribonucleoproteins (snRNP). There are 117 identified snRNA genes in C. elegans.
There are 612 nuclear and 22 mitochondrial transfer RNA (tRNA) genes in C. elegans. Seven nuclear genes have been identified as suppressor tRNAs: sup-5 (B0523.t1), sup-7 (C03B1.t1), sup-21 (C06E2.t1), sup-24 (Y73F8A.t2), sup-29 (Y37E11AL.t1), sup-28 (C48C5.t6), and sup-33 (D1073.t1). Two are likely to be pseudogenes: rtw-5 (K06G5.t1), and rtw-6 (Y40C5A.t1). The remainder were predicted computationally (Lowe and Eddy, 1997). The tRNA genes range in size from 64 to 122 bp with 72% having 72 or 73 bp. 29 of the 608 are genes with two exons and the remainder have a single exon. tRNAscan also predicts that there are 213 tRNA pseudogenes. Nuclear tRNA genes are over-represented on the X chromosome with 45% residing there. The other 55% are distributed uniformly over the autosomes; however, there is a slight enrichment on chromosome III and a lower density in the central region of chromosome IV and on the left arm of chromosome I.
Approximately 70% of C. elegans mRNAs are covalently modified at their 5’ end by the addition of 22-nt trans-spliced leader RNA sequences. C. elegans contains 128 trans-spliced leader (TSL) RNA genes which encode 2 sub-types of TSL. The most abundant form, SL1, is mainly trans-spliced to the 5’ end of pre-mRNAs, including the first cistron in polycistronic (operon) pre-mRNAs; the rarer form, SL2, is generally trans-spliced to downstream cistrons in polycistronic operons. The C. elegans genome contains 110 SL1 RNA genes on the 1 kb tandem repeat that also contains the genes for 5S rRNA (Krause and Hirsh, 1987). In contrast, the genome contains only 18 dispersed SL2 RNA genes, which specify a variety of variant SL2 RNAs (Stein et al., 2003). Some of these have different SL2 sequences, and in the past these have been given different names, such as SL3, SL4, etc. (Ross et al., 1995). Nonetheless, they are all variants of SL2 and now are given the names SL2a, SL2b, etc. They are used randomly at SL2-accepting trans-splice sites (T. Blumenthal, unpublished). The C. briggsae genome also contains 18 SL2 RNA genes, and all 36 genes from the two species descended from four primordial SL2 RNA genes present in their last common ancestor (Stein et al., 2003). Trans-splicing is covered in more detail in Trans-splicing and operons in C. elegans.
C. elegans chromosomes have several distinctive features. Instead of having centromeres embedded in highly repeated sequences, its chromosomes are holocentric, with microtubule attachment sites distributed along their length. In hermaphrodites (XX), gene expression from both X chromosomes is down-regulated in somatic cells by a dosage compensation mechanism in order to approximately match the expression in males, which have one X chromosome (XO) (See X-Chromosome Dosage Compensation). C. elegans autosomes have distinct domains—a central region flanked by two distal “arms” that together comprise more than half of the chromosome (Table 1). Compared with the centers, the arms have higher meiotic recombination rates, lower gene density, and higher repeat content (Rockman et al., 2009; The C. elegans Sequencing Consortium 1998; Barnes et al., 1995). Arms are not as sharply defined on the X chromosome.
Protein-coding genes are found equally on either strand of DNA and are fairly uniformly distributed throughout the genome. They are slightly denser on autosomes than on chromosome X (Table 1) and, in general, the central regions of the autosomes are denser than the arms. The left arm of chromosome II is an exception.
Genes in general do not overlap one another, that is to say, their coding exons do not overlap, but there are numerous examples of genes that fall within introns of another gene, either on the same or the opposite strand. F10F2.2 contains 5 coding genes on the opposite strand in 2 large introns, in addition to two ncRNA genes in the same sense. There are examples of pairs of transcripts having non-overlapping coding exons but sharing the same locus, for example, unc-17 (ZC416.8) and cha-1 (ZC416.8). These two genes share a common promoter and a first, non-coding exon. The rest of the coding exons do not overlap, so the two genes encode different proteins with mutationally separable functions: unc-17 encodes a synaptic vesicle acetylcholine transporter, while cha-1 encodes a choline acetyltransferase (Rand et al. 2000; Alfonso et al., 1994). Other examples of non-overlapping transcripts sharing one locus include lev-10 (Y105E8A.7) and eat-18 (Y105E8A.7), wars-2 (C34E10.4b) and prx-10 (C34E10.4a), and Y65B4BL.1a and Y65B4BL.1b.
Most genes are transcribed from a single sense strand of the locus. An exception is the pair of loci, eri-6 (C41D11.1) and eri-7 (C41D11.7), that are in adjacent locations but opposite strands. They produce separate pre-messenger RNAs (pre-mRNAs) which are trans-spliced to form a functional mRNA, eri-6/7. Trans-splicing of eri-6/7 is mediated by a direct repeat that flanks the eri-6 gene (Fischer et al. 2008). Another unusual locus is unc-49 (T21C12.1), which contains a single copy of a GABA receptor N terminus, followed by three tandem copies of a GABA receptor C terminus. Using a single promoter, unc-49 generates three distinct GABA-A receptor-like subunits by splicing the N terminus to each of the three C-terminal repeats (Bamber et al. 1999).
Examples of RNA editing in C. elegans are rare. adr-1 (H15N14.1) and adr-2 (T20H4.4) have been shown to mediate editing of the RNA products of ZC239.6, exos-4.2 (Y6D11A.1), unc-64 (F56A8.7a), W03D8.2, and lam-2 (C54D1.5) in nervous, and possibly vulval, tissue (Tonkin et al. 2002). There is also C-to-U RNA editing in gld-2 (ZC308.1) transcripts in germline cells, although somatic cell gld-2 transcripts show no detectable editing. (Wang et al. 2004).
A characteristic feature of the worm genome is the existence of genes organized into operons. These polycistronic gene clusters contain two or more closely spaced genes, which are oriented in a head to tail direction. They are transcribed as a single polycistronic mRNA and separated into individual mRNAs by the process of trans-splicing (Spieth et al., 1993). These topics are covered in detail in Trans-splicing and operons in C. elegans.
This chapter has described some of the ways in which gene structures in WormBase are curated. Inevitably there will be new technologies which will be developed in the future that have greater or lesser effects on the way the structure of genes can be defined and understood. WormBase curators will continue to endeavor to use all available data to refine and correct the structures of genes, and to review our curation policies such as when to add a rarely-observed alternate splicing event to the set of isoforms at a locus, how much effort to put into manually curating ncRNA genes and how much attention should be given to curating transcripts that are antisense to a coding gene. Many questions that have some bearing on the structure of genes also remain to be addressed, such as how alternately spliced transcripts at a locus are regulated and the precise definition of promoters, enhancers and other regulatory regions.
This work was supported by grants from the National Human Genome Research Institute (#U41 HG002042, principal investigator, R.K.W.; and P41-HG002223, principal investigator, P.W.S.) and the British Medical Research Council.
Alfonso, A., Grundahl, K., McManus, J.R., Asbury, J.M., and Rand, J.B. (1994). Alternative splicing leads to two cholinergic proteins in Caenorhabditis elegans. J. Mol. Biol. 24, 627-630. Abstract Article
Bamber, B.A., Beg, A.A., Twyman, R.E., and Jorgensen, E.M. (1999). The Caenorhabditis elegans unc-49 locus encodes multiple subunits of a heteromultimeric GABA receptor. J. Neurosci. 19, 5348-5359. Abstract
Barnes, T.M., Kohara, Y., Coulson, A., and Hekimi, S. (1995). Meiotic recombination, noncoding DNA and genomic organization in Caenorhabditis elegans. Genetics. 141, 159-179. Abstract
Batista, P.J., Ruby, J.G., Claycomb, J.M., Chiang, R., Fahlgren, N., Kasschau, K.D., Chaves, D.A., Gu, W., Vasale, J.J., Duan, S., et al. (2008). PRG-1 and 21U-RNAs interact to form the piRNA complex required for fertility in C. elegans. Mol. Cell. 31, 67-78. Abstract Article
Blumenthal T., Trans-splicing and operons in C. elegans (November 20, 2012). WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.5.2, http://www.wormbook.org.
Blumenthal, T., and Steward, K. (1997). RNA processing and gene structure. In: C. elegans II, D.L. Riddle, T. Blumenthal, B.J. Meyer, J.R. Priess, eds. (Plainview, New York: Cold Spring Harbor Laboratory Press), pp. 117-145.
Bracht, J., Hunter, S., Eachus, R., Weeks, P., and Pasquinelli, A.E. (2004). Trans-splicing and polyadenylation of let-7 microRNA primary transcripts. RNA 10, 1586-1594. Abstract Article
The C. elegans Sequencing Consortium (1998). Genome sequence of the nematode C. elegans: A platform for investigating biology. Science 282, 2012-2018. Abstract Article
Castillo-Davis, C.I., Mekhedov, S.L., Hartl, D.L., Koonin, E.V., and Kondrashov, F.A. (2002). Selection for short introns in highly expressed genes. Nat. Genet. 31, 415-418. Abstract
Chen, R.A., Down, T.A., Stempor, P., Chen, Q.B., Egelhofer, T.A., Hillier, L.W., Jeffers T.E., and Ahringer J. (2013). The landscape of RNA polymerase II transcription initiation in C. elegans reveals promoter and enhancer architectures. Genome Res. 23, 1339-1347. Abstract Article
Ellis, R.E., Sulston, J.E., and Coulson, A.R. (1986). The rDNA of C. elegans: sequence and structure. Nucleic Acids Res. 11, 2345-2364. Abstract Article
Galagan, J.E., Calvo, S.E., Borkovich, K.A., Selker, E.U., Read, N.D., Jaffe, D., FitzHugh, W., Ma, L.J., Smirnov, S., Purcell, S., et al. (2003). The genome sequence of the filamentous fungus Neurospora crassa. Nature 422, 859-868. Abstract Article
Fischer, S.E., Butler, M.D., Pan, Q., and Ruvkin G. (2008). Trans-splicing in C. elegans generates the negative RNAi regulator ERI-6/7. Nature 455, 491-496. Abstract Article
Gerstein, M.B., Bruce C., Rozowsky J.S., Zheng, D., Du, J., Korbel, J.O., Emanuelsson, O., Zhang, Z.D., Weissman, S., and Snyder M. (2007). What is a gene, post-ENCODE? History and updated definition. Genome Res. 17, 669-681. Abstract Article
Gerstein M.B., Lu Z.J., Van Nostrand E.L., Cheng C., Arshinoff B.I., Liu, T., Yip, K.Y., Robilotto, R., Rechsteiner, A., Ikegami, K., et al. (2010). Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 330, 1775-1787. Abstract Article
Gu, W., Lee, H.C., Chaves, D., Youngman, E.M., Pazour, G.J., Conte, D. Jr., and Mello C.C. (2012). CapSeq and CIP-TAP identify Pol II start sites and reveal capped small RNAs as C. elegans piRNA precursors. Cell 151, 1488-1500. Abstract Article
Harrison, P.M., Echols, N., and Gerstein, M.B. (2001). Digging for dead genes: an analysis of the characteristics of the pseudogene population in the Caenorhabditis elegans genome. Nucleic Acids Res. 29, 818-830. Abstract Article
Heschl, M.F., and Baillie, D.L. (1989). Identification of a heat-shock pseudogene from Caenorhabditis elegans. Genome 32, 190-1955. Abstract Article
Hwang, B.J., Müller, H.M., Sternberg, P.W. (2004). Genome annotation by high-throughput 5’ RNA end determination. Proc. Natl. Acad. Sci. U. S. A. 101, 1650-1655. Abstract Article
The International Human Genome Sequencing Consortium. (2001). Initial sequencing and analysis of the human genome. Nature 409, 860-921. Abstract Article
Jan, C.H., Friedman, R.C., Ruby, J.G., and Bartel, D.P. (2011). Formation, regulation and evolution of Caenorhabditis elegans 3'UTRs. Nature 469, 97-101. Abstract Article
Kozomara, A., and Griffiths-Jones, S. (2011). miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 39(Database Issue), D152-D157. Abstract Article
Krause, M., and Hirsh, D. (1987). A trans-spliced leader sequence on actin mRNA in C. elegans. Cell 49, 753-761. Abstract Article
Kruesi, W.S., Core, L.J., Waters, C.T., Lis, J.T., and Meyer, B.J. (2013). Condensin controls recruitment of RNA polymerase II to achieve nematode X-chromosome dosage compensation. Elife 2, e00808. Abstract Article
Lamm, A.T., Stadler, M.R., Zhang, H., Gent, J.I., and Fire A.Z. (2011). Multimodal RNA-seq using single-strand, double-strand, and CircLigase-based capture yields a refined and extended description of the C. elegans transcriptome. Genome Res. 21, 265-275. Abstract Article
Loftus, B.J., Fung, E., Roncaglia, P., Rowley, D., Amedeo, P., Bruno, D., Vamathevan, J., Miranda, M., Anderson, I.J., Fraser, J.A., et al. (2005). The genome of the basidiomycetous yeast and human pathogen Cryptococcus neoformans. Science 307, 1321-1324. Abstract Article
Lowe, T.M., and Eddy, S.R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955-964. Abstract Article
Mangone, M., Manoharan, A.P., Thierry-Mieg, D., Thierry-Mieg, J., Han, T., Mackowiak, S.D., Mis, E., Zegar, C., Gutwein, M.R., Khivansara, V., et al. (2010). The landscape of C. elegans 3'UTRs. Science 329, 432-435. Abstract Article
Meyer, B.J. X-Chromosome dosage compensation (June 25, 2005). WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.8.1, http://www.wormbook.org.
Mounsey, A., Bauer, P., and Hope, I.A. (2002). Evidence suggesting that a fifth of annotated Caenorhabditis elegans genes may be pseudogenes. Genome Res. 12, 770-775. Abstract Article
Nam, J.W., and Bartel, D. (2012). Long non-coding RNAs in C. elegans. Genome Res. 22, 2529-2540. Abstract Article
Nelson, D.W., and Honda, B.M. (1985). Genes coding for 5S ribosomal RNA of the nematode Caenorhabditis elegans. Gene 38, 245-251. Abstract Article
Nowrousian, M., Wurtz, C., Poggeler, S., and Kuck, U. (2004). Comparative sequence analysis of Sordaria macrospora and Neurospora crassa as a means to improve genome annotation. Fungal Genet. Biol. 41, 285-292. Abstract Article
Park, J.H., Ahn, S., Kim, S., Lee, J., Nam, J.W., and Shin, C. (2013). Degradome sequencing reveals an endogenous microRNA target in C. elegans. FEBS Lett. 587, 964-969. Abstract Article
Prachumwat, A., DeVincentis, L., and Palopoli, M.F. (2004). Intron size correlates positively with recombination rate in Caenorhabditis elegans. Genetics 163, 1585-1590. Abstract Article
Rand, J.B, Duerr, J.S., and Frisby, D.L. (2000). Neurogenetics of vesicular transporters in C. elegans. FASEB J. 15, 2414-2422. Abstract Article
Reboul, J., Vaglio, P., Rual, J.F., Lamesch, P., Martinez, M., Armstrong, C.M., Li, S., Jacotot, L., Bertin, N., Janky, R., et al. (2003). C. elegans ORFeome version 1.1: experimental verification of the genome annotation and resource for proteome-scale protein expression. Nat. Genet. 34, 35-41. Abstract Article
Robertson, H.M. (1998). Two large families of chemoreceptor genes in the nematodes Caenorhabditis elegans and Caenorhabditis briggsae reveal extensive gene duplication, diversification, movement, and intron loss. Genome Res. 8, 449-463. Abstract
Robertson, H.M. (2000). The large srh family of chemoreceptor genes in Caenorhabditis nematodes reveals processes of genome evolution involving large duplications and deletions and intron gains and losses. Genome Res. 10, 192-203. Abstract Article
Robertson, H.M. (2002). Updating the str and srj (stl) families of chemoreceptors in Caenorhabditis nematodes reveals frequent gene movement within and between chromosomes. Chem. Senses 26, 151–159. doi: 10.1093/chemse/26.2.151 Article
Robertson, H.M., and Thomas, J.H. The putative chemoreceptor families of C. elegans (January 06, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.66.1, http://www.wormbook.org.
Rockman M.V., and Krulyak L. (2009). Recombinational landscape and population genomics of Caenorhabditis elegans. PLoS Genet. 5, e1000419. Abstract Article
Rosenzweig B., Liao L.W., and Hirsh D. (1983). Sequence of the C. elegans transposable element Tc1. Nucleic Acids Res. 11, 4201–4209. Abstract Article
Ross, L.H., Freedman, J.H., and Rubin, C.S. (1995). Structure and expression of novel spliced leader RNA genes in Caenorhabditis elegans. J. Biol. Chem. 270, 22066–22075. Abstract Article
Ruby, J., Jan, C., and Bartel, D.P. (2007). Intronic microRNA precursors that bypass Drosha processing. Nature 448, 83-86. Abstract Article
Ruby, J. Jan, C., Player, C. Axtell, M., Lee, W., Nusbaum, C., Ge, H., and Bartel, D. (2006). Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell 127, 1193-1207. Abstract Article
Saito, T.L., Hashimoto, S.I., Gu, S.G., Morton, J.J., Stadler, M., Blumenthal ,T., Fire, A., and Morishita S. (2013). The transcription start site landscape of C. elegans. Genome Res. 23, 1348-1361. Abstract Article
Sibley, M.H., Johnson, J.J., Mello, C.C., and Kramer, J.M. (1993). Genetic indentification, sequence, and alternative splicing of the Caenorhabditis elegans α2(IV) collagen gene. J. Cell Biol. 123, 255-264. Abstract
Snyder, M., and Gerstein, M. (2003). Genomics. Defining genes in the genomics era. Science 300, 258–260. Abstract Article
Spieth, J., Brooke, G., Kuersten, S., Lea, K., and Blumenthal, T. (1993). Operons in C. elegans: polycistronic mRNA precursors are processed by trans-splicing of SL2 to downstream coding regions. Cell 73, 521–532. Abstract Article
Stein, L.D., Bao, Z., Blasiar, D., Blumenthal, T., Brent, M.R., Chen, N., Chinwalla, A., Clarke, L., Clee, C., Coghlan, A., et al. (2003). The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol. 1, 166–192. Abstract Article
Sulston, J.E. and Brenner, S. (1974). The DNA of Caenorhabditis elegans. Genetics 77, 95-104. Abstract
Tonkin, L.A., Saccomanno, L., Morse, P.M., Brodigan, T., Krause, M., and Bass, B.L. (2002). RNA editing by ADARs is important for normal behavior in Caenorhabditis elegans. EMBO J. 21, 6025–6035. Abstract Article
Vaglio, P., Lamesch, P., Reboul, J., Rual, J.F., Martinez, M., Hill, D., and Vidal, M. (2003). WorfDB: the Caenorhabditis elegans ORFeome Database. Nucleic Acids Res. 31, 237–240. Abstract Article
Van Horn, D.J., Eisenberg, D., O’Brien, C.A., and Wolin, S.L. (1995). Caenorhabditis elegans embryos contain only one major species of Ro RNP. RNA 1, 293-303. Abstract
Wang, L., Kimble, J., and Wickens, M. (2004). Tissue-specific modification of gld-2 mRNA in C. elegans: likely C-to-U editing. RNA. 10, 1444-1448. Abstract Article
Ward, S., Burke, D.J., Sulston, J.E., Coulson, A.R., Albertson, D.G., Ammons, D., Klass, M., and Hogan, E. (1988). Genomic organization of major sperm protein genes and pseudogenes in the nematode Caenorhabditis elegans. J. Mol. Biol.199, 1–13. Abstract Article
Williams, G.W., Davis, P.A., Rogers, A.S., Bieri, T., Ozersky P., and Spieth, J. (2011). Methods and strategies for gene structure curation in WormBase. Database 2011, baq039. Abstract Article
Zahler, A.M. Pre-mRNA splicing and its regulation in Caenorhabditis elegans (March 21, 2012). WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.31.2, http://www.wormbook.org.
Zhang, H., and Emmons, S.W. (2000). A mediator protein confers regulatory selectivity on lineage-specific expression of a transcription factor gene. Genes Dev. 14, 2161–2172. Abstract Article
*Edited by Jonathan Hodgkin. Last revised. Published October 29, 2014. This chapter should be cited as: Spieth et al. Overview of gene structure in C. elegans (October 29, 2014), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.65.2, http://www.wormbook.org.
Copyright: © 2014 John Spieth, Daniel Lawson, Paul Davis, Gary Williams, Kevin Howe. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: [email protected]
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.