Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
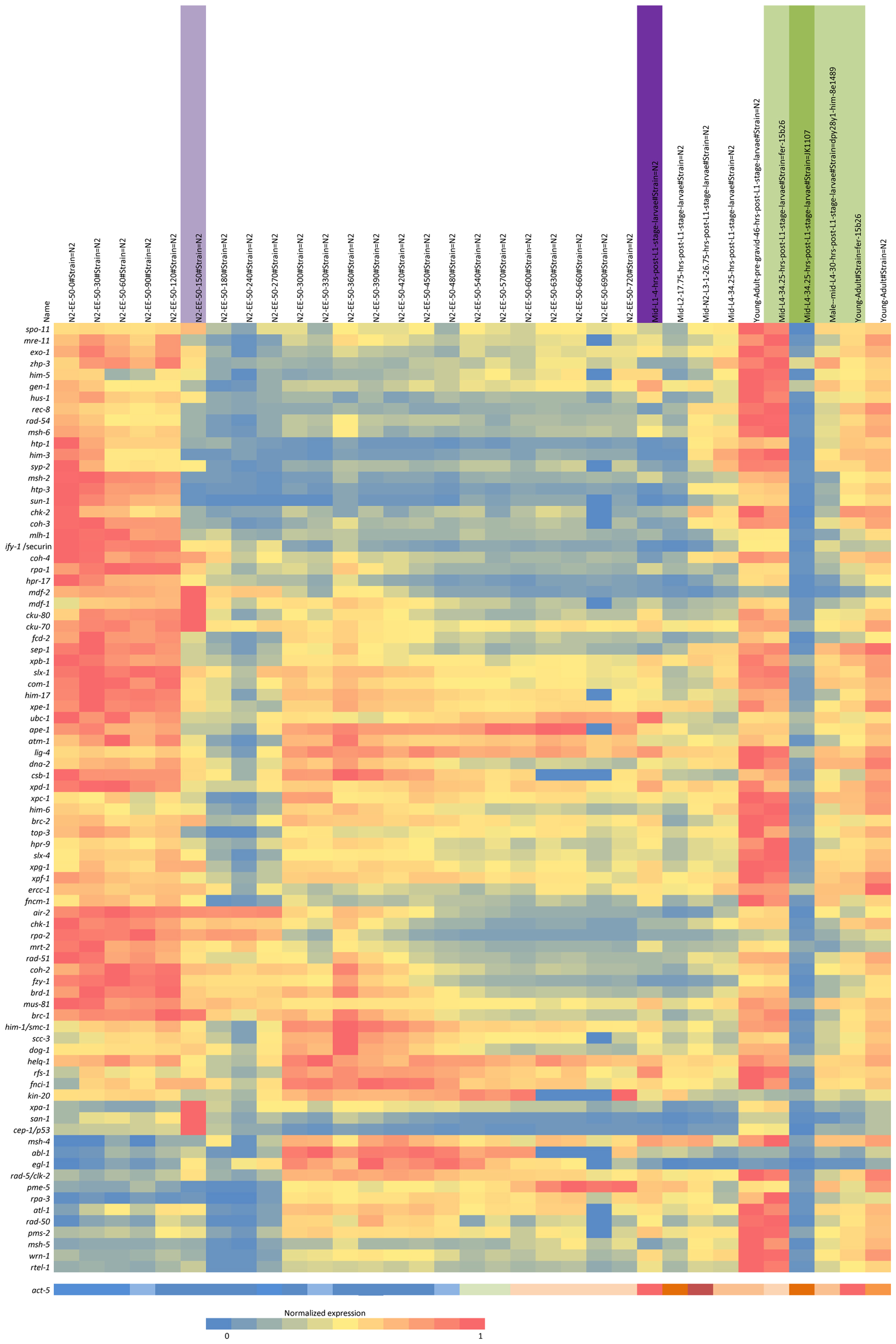
Integrity of genetic information is essential to both individual health and reproductive capacity. The Caenorhabditis elegans model system provides an opportunity not only to investigate the components of the various repair pathways and the pathway interactions, but also to study the role of repair in different developmental stages. A number of different DNA repair pathways respond to damage dependent upon the nature of the damage, type of cell, stage of development, and genomic location, for example, at the telomeres (Astin et al., 2008; Clejan et al., 2006; reviewed in Lans and Vermeulen, 2011). It is well known, for example, that the response to DNA double strand breaks (DSBs) in the proliferating premeiotic germline differs from that during meiosis or embryonic cleavage, and from somatic cell replication (Vermezovic et al., 2012; Couteau and Zetka, 2011; Lans et al., 2010; McLellan et al., 2009; Pontier and Tijsterman, 2009; Lee et al., 2007; Holway et al., 2006; Weidhaas et al., 2006). For example, differences between somatic and germ line cell response to DNA damage were examined and molecular mechanisms for somatic cell resistance to DNA damage-induced cell death described (Vermezovic et al., 2012). As an animal system, C. elegans provides a useful model to more fully understand the integration of the types of repair that occur in response to DNA damage in different cell types during growth and development (See the developmental profile of repair gene RNA in Figure 1).
|
Figure 1. Expression profile during C. elegans development. Expression profile clustering of a set of DNA repair genes using nearest neighbor hierarchical clustering of the normalized RNA-seq data from the modEncode website is shown (www.modencode.org). The cluster analysis has been modified by hand somewhat to aggregate some groupings. The top axis is the developmental profile from fertilization to adult hermaphrodite. There is a clear change in the expression pattern at column six at the onset of gastrulation (150 min post fertilization), marked by a purple bar. A darker purple bar marks the mid-first larval stage. The green bars indicate mutant strains used. The darker green is a germline-minus strain, JK1107. Adjacent to the left are mid-larval females and young pre-gravid adult hermaphrodites. Clustering and figure prepared by Dr. Jeffrey Chu, University of British Columbia.
Since the previous version of this chapter (DNA repair), there has been an explosion of research using C. elegans to study many aspects of DNA repair and several reviews have been published (Table 1). In this chapter, the focus will be on research in the past few years beginning with the use of C. elegans in the study of the Fanconi Anemia (FA) repair pathway (reviewed in Youds et al., 2009; see also Jones and Rose, 2012). The FA pathway (see below) is involved in identifying replication blocks that can result from either unresolved DNA secondary structures (Kruisselbrink et al., 2008; Youds et al., 2006; Cheung et al., 2002) or from interstrand crosslinks (ICLs) (reviewed in McVey, 2010; Youds et al., 2009; and more generally in Kottemann and Smogorzewska, 2013; Deans and West, 2011). Repair involves translesion synthesis (TLS) and homologous recombination (HR) repair but not nonhomologous endjoining (NHEJ) (Youds et al., 2006). A key component of the FA pathway, FCD-2/FANCD2, is involved in directing repair towards error-free HR and away from error-prone NHEJ. This important choice has been shown to be under genetic control and regulated by FANCD2, which binds to DNA lesions and recruits repair proteins. In C. elegans, genetic interaction analysis demonstrated that elimination of NHEJ by inhibition of LIG4 suppressed the repair defects of Fcd-2 mutants (Adamo et al., 2010). In both C. elegans and human cells (Adamo et al., 2010; Pace et al., 2010; reviewed in Bunting and Nussensweig, 2010), it was discovered that FANCD2 is required to prevent NHEJ, which can lead to chromosomal rearrangements that may be mutagenic and toxic to the cell. HR repair, on the other hand, is essentially error free and thus the more desirable pathway for faithful reproduction.
Table 1: Reviews cited
| Topic | Reference |
|---|---|
| Fanconi anemia and ICL repair | |
| FA Repair in avian cells | Takata et al., 2006 |
| DOG-1/FANCJ and G4 DNA repair | Maizels, 2008; Brosh, 2011 |
| G4 DNA and human disease | Wu and Brosh, 2010 |
| Interstrand crosslink repair | Kottemann and Smogorzewska, 2013; Deans and West, 2011; McVey, 2010 |
| Chromatin modification at DSB | Fischle, 2009 |
| Double strand break repair | Lemmens and Tijsterman, 2011; Pontier and Tijsterma, 2009 |
| Phosphorylation in DSB repair | Summers et al., 2011 |
| Cohesion and DNA repair | Sjögren and Strom, 2009; Watrin and Peters, 2006 |
| C. elegans cohesins | Wood et al., 2010 |
| PARPs and synthetic lethality | St-Laurent and Desnoyers, 2011; Helleday, 2011 |
| Nucleotide excision repair | Lans and Vermeulen, 2011 |
Table 2: Known components of the FA pathway in C. elegans
| Component | Gene Function | Reference (also www.wormbase.org) |
|---|---|---|
| BRC-2 | Loads RAD-51 | Ko et al., 2008; Petalcorin et al., 2007; Martin et al., 2005 |
| FCD-2 | Promotes HR repair | Adamo et al., 2010; Lee et al., 2007; Collis et al., 2006 |
| FNCI-1 | Required for FCD-2 focus formation | Lee, K.Y. et al., 2010 |
| DOG-1 | Unwinds G-tracts | Youds et al., 2008; Youds et al., 2006; Cheung et al., 2002 |
| FNCM-1 | Required for ubiquitination of FCD-2 | Lee, K.Y. et al., 2010 |
| RFS-1 | RAD-51, paralog in HR repair | Yanowitz, 2008; Ward et al., 2007 |
| SLX-4 | endonuclease in HR repair | Saito et al., 2009 |
The Fanconi anemia pathway is so-named because of a genetic disease in humans first described by G. Fanconi (1927, in Lobitz and Velleuer, 2006). In humans, FA is a rare recessive genetic disorder characterized by bone marrow failure resulting in anemia and accompanied by developmental abnormalities and cancer susceptibility as a result of subsequent genome instability (www.fanconi.org). Diagnosis involves cellular hypersensitivity to DNA crosslinking agents and more recently whole genome sequencing (WGS) (Ameziane et al., 2012). The pathway consists of a core complex of proteins that mono-ubiquinate FANC D2 and I, which then localize to sites of DNA-damage and promote HR repair. There are currently 15 genetic subtypes that have been described (A, B, C, D1 [BRCA2], D2, E, F, G, I, J, L, M, N, O and P) (Ameziane et al., 2012; reviewed in Kottemann and Smogorzewska, 2013; Deans and West, 2011). In C. elegans as in other species, the FA pathway is involved in identifying replication blocks and initiating subsequent HR repair (Ward et al., 2007). Seven C. elegans FA protein orthologs have been defined by knockout mutations in the genes coding for them: D1—brc-2(tm1086), D2—fcd-2(tm1298), I—fnci-1 (tm3148), J—dog-1(gk10), M—fncm-1(tm3081), O—him-15/rfs-1(ok2702) a RAD51C ortholog, and P—him-18/slx-4(tm2181). Orthologs of the remaining FA components identified in humans, are not immediately recognizable by DNA sequence matches and if present will need to be identified by other means.
Research into the Fanconi anemia pathway in C. elegans began with the discovery that a member of the iron-sulfur class of helicases, DOG-1 was involved in unwinding long (17–27 bp) stretches of poly-G/poly-C DNA (Cheung et al., 2002). In the absence of DOG-1, small deletions detectable by PCR were observed. The deletions initiated within poly-G/poly-C DNA and extended for variable lengths upstream, consistent with the proposal that the G-tracts formed secondary structures blocking replication on the lagging strand. The number of deletions generated in the absence of DOG-1 was increased in the absence of translesion synthesis (TLS) or homologous recombination (HR) repair, but not nonhomologous endjoining (NHEJ) (Youds et al., 2006). Subsequently it was demonstrated that DOG-1 is orthologous to FANCJ (Youds et al., 2008) and hence the connection with the FA repair pathway was confirmed. In humans, it was known that the breast cancer-interacting protein (BRIP)/breast cancer associated helicase (BACH1)/FANCJ gene was connected to HR repair (Cantor et al., 2001). Thus, to some degree the functional relationship between DOG-1/FANCJ and HR repair is conserved. However, FANCJ in humans contains a breast cancer interacting domain (Cantor et al., 2001) that is absent in chicken (Takata et al., 2006) and C. elegans (Youds et al., 2008). The domain is not required for resolution of secondary G-tract structures as shown by the results from C. elegans. The fact that neither the worm nor the chicken have the BRCA-1 interacting domain and yet both these species utilize HR repair following lesions caused by the absence of their respective FANCJ orthologs (Kitao et al., 2011; Takata et al., 2006; Youds et al., 2006) indicates that the presence of this domain is not essential for recruiting HR repair. Thus, examination of DOG-1 activity may help to clarify the relationship of DOG-1 to HR repair and the degree of overlap with other pathways that result in HR repair, such as those involving BRC-1.
Covalent linkage of DNA, either inter- or intra-strand, can occur as a result of exposure to endogenous or exogenous crosslinking agents. The presence of an ICL is especially problematic for DNA replication and needs to be removed to prevent blockage. In general, repair of ICLs involves the FA pathway and requires HR and TLS (reviewed in Deans and West, 2011). In C. elegans, treatment with ICL agents, if unrepaired, can result in checkpoint-induced cell cycle arrest, DNA damage such as chromatin bridges and breaks, or apoptosis of germ cells (Lee, S.J. et al., 2010; Youds et al., 2008). Untimely repair can result in developmental delays and improperly repaired damage can lead to chromosomal rearrangements, whereas unrepaired ICLs can result in cell death, reduced fecundity in the adult, and embryonic lethality of the progeny.
During ICL repair both TLS and HR are involved. TLS polymerases are specialized DNA polymerases that insert nucleotides at the site of lesions with reduced fidelity (Roerink et al., 2012; Kim and Michael, 2008;). In C. elegans, the relationship between these two types of repair has been clarified to some extent by the characterization of two gene products, POLQ-1 and HEL-308, which are proposed to function in different DNA repair pathways (Muzzini et al., 2008; reviewed in McVey, 2010). POLQ-1 has a helicase domain at the N-terminus and a polymerase domain at the C-terminus, and in other species has been implicated in recombination-independent and TLS-dependent ICL repair. In C. elegans, POLQ-1 appears to function in a pathway with BRC-1 that is different than the pathway involving HEL-308, FNCD-2, and DOG-1 (reviewed in Youds et al., 2008). In addition to DOG-1/FANCJ (Youds et al., 2008), absence of additional FA pathway components FANCM and FANCI (Lee K.Y. et al., 2010), and the FANCD2-associated nuclease FAN1 (Kratz et al., 2010; Liu et al., 2010; MacKay et al., 2010; Smogorzewska et al., 2010) results in sensitivity to ICL-inducing agents, such as UVA-activated trimethylpsoralen, nitrogen mustard, and cisplatin, but not DNA-breakage agents such as X-rays or UVC.
With regard to ICL sensitivity, loss of DOG-1 function has the same phenotype, which is measured by the number of unhatched embryos, as does loss of both DOG-1 and FCD-2. The phenotype is not due to double-strand break (DSB) repair because DOG-1 loss of function does not confer sensitivity to radiation-induced DSBs. Furthermore, DOG-1 is not required for the formation of RAD-51 nor FCD-2 foci, indicating that it is not responsible for transmitting the signal that a block has occurred (Youds et al., 2008). So, what is its role in ICL sensitivity? In the absence of DOG-1, there are increased numbers of PCR-detected deletions in a FCD-2 deficient background, indicating that in the absence of DOG-1, FCD-2-initiated repair is involved at replication blocks caused by unresolved secondary structures (Youds et al., 2008; Youds et al., 2006). Is the role of DOG-1 in ICL repair and in resolving secondary structures blocking replications the same? The fact that mutants lacking DOG-1 and FCD-2 have increased deletions, but not increased embryonic lethality may appear contradictory. However, taken in the context of secondary structures resolution, DOG-1’s absence would produce structures that can not only form replication blocks but that can also become substrate for crosslinking (either extraneous or endogenous). In the latter case, absence of DOG-1 may generate an ICL-sensitive phenotype not because of its role in repairing crosslinks, but rather as a result of unresolved structures that potentiate crosslinking. In this perspective DOG-1’s function would be the same in both, and outside the FCD-2 pathway (Jones and Rose, 2012).
In the absence of DOG-1/FANCJ, unresolved secondary structures also (in addition to PCR-detectable deletions and ICL sensitivity) lead to chromosomal rearrangements, deletions, and translocations (Zhao et al., 2008). Those analyzed have G-rich or G-tract sequences at their breakpoints. The rearrangements could result from improper repair at replication blocks involving NHEJ and the formation of inappropriately joined chromosomal fragments. However, since some of the rearrangements involve two different chromosomes, it seems possible that these cases involve ectopic interactions between G-tracts on heterologous chromosomes. DNA breaks may occur and result in translocated chromosomes either through NHEJ of broken ends or by breakage-induced repair from the broken end using the sister as template. In C. elegans there are nearly 400 copies of DNA sequences containing 18 or more consecutive G's, which are substrate for secondary structure formation. These G-tracts are dispersed across the chromosomes and their number and distribution, but not position, are conserved with C. briggsae (Zhao et al., 2007). The number of C/G-tracts is much greater than would be expected by chance and greater than the number of A/T tracts (Denver et al., 2004). The fact that they are retained despite their potential obstruction to replication, that there is a mechanism for their removal (loss by deletion), and that they are conserved between species may indicate a biological role for G-tract secondary structures. One such role in facilitating associations between homologs during meiosis has been proposed by Sen and Gilbert (1988). A review of the biology of G-quadraplexes is presented in Brosh (2011). Regardless of their potential biological roles, tetra-G complexes have been proposed a target for small molecule intervention in humans (Rodriguez et al., 2012).
At critical times when cells progress from one stage to the next, the quality of the DNA is assessed by cell cycle checkpoints. If the chromosomes don't pass quality control, the checkpoint may generate a ‘Wait’ signal. A classic example is the metaphase to anaphase checkpoint, MAD-2, first describe in yeast (Li and Murray, 1991) and found to be functionally orthologous in C. elegans (Kitagawa and Rose, 1999). In the absence of the checkpoint, if cells progress to the next stage, the premature progression can result in mitotic segregation defects in the germline (Kitagawa and Rose, 1999) or the embryo (Encalada et al., 2005). A different checkpoint protects the cell from DNA damage. In the germline, damage can lead to apoptosis (nonprogrammed cell death), cell cycle arrest, or heritable mutation. The C. elegans ortholog of the transcription factor p53, CEP-1, which is a key regulator of the DNA damage checkpoint, is required for DNA damage-induced apoptosis in the C. elegans germline, but not for programmed cell death occurring during worm development nor physiological (radiation-independent) germ cell death (Jaramillo-Lambert et al., 2010; Derry et al., 2001; Schumacher et al., 2001). The absence of checkpoints in the germline can result in a range of problems leading to chromosomal and genome instability.
Many DNA repair pathways, including DSB, NHEJ, and the DNA damage response (DDR), rely on checkpoints such as ATM, ATR, and DNA-PK kinases (reviewed in Lemmens and Tijsterman, 2011; Summers et al., 2011). In the case of replication block, ubiquitination of FANCD2 and focus formation require the checkpoint proteins, RPA, ATR, and CHK1 (Lee, K.Y. et al., 2010). It is not completely understood how the decision to follow one repair pathway versus another is made, but it is well known that members of the phosphatidylinositol 3-kinase related kinase (PIKK) family are primary sensors of DNA damage. In C. elegans, ATM (ataxia-telangiectasis mutated) and ATR (ATM and Rad3-related) may perform independent functions in distinct DNA damage checkpoint signaling pathways, with ATR signaling at replication blocks (Garcia-Muse and Boulton, 2005) and ATM at DSBs (Jones et al., 2012). In addition to signaling in response to ionizing radiation, ATM is required for genomic stability and is its absence is a spontaneous mutator (Jones et al., 2012).
Upstream of ATM and ATR the Werner's Syndrome helicase WRN-1 functions in response to DSBs and replication inhibition. WRN-1 is a 3’ to 5’ helicase capable of unwinding a number of DNA structures and its absence results in an increased sensitivity to DNA damage (Hyun et al., 2008 ). During proliferation of the mitotic cells, the pathway leading to arrest or apoptotic cell death following UV-C treatment has been described (Stergiou et al., 2007). This pathway overlaps with, but is distinct from the pathway activated by ionizing radiation. UV-C-induced lesions are recognized and processed by the nucleotide excision repair pathway and some may apparently become DSBs, which are then processed by homologous recombination (Stergiou et al., 2011).
DSBs can occur as a consequence of replication blocks, damage by ionizing radiation, or as genetically regulated initiators of meiotic crossing over. Central to DSB repair is the RecA homolog, Rad51, which mediates functions involving strand invasion during recombination, and the nuclease MRE-11 (Chin and Villeneuve, 2001). A number of well-conserved repair pathways and regulators of DSBs exist and maintain genome stability when breaks occur. The network of DSB repair, including meiotic crossing over, has been recently reviewed in depth (Lemmens and Tijsterman, 2011). With regard to replication and repair, DSBs can be repaired by either NHEJ or HR repair. The regulation of which pathway is involved occurs in the context of the source of damage, the timing in the cell cycle, and the stage of development. In C. elegans, HR is used to repair radiation-induced DSBs in cycling somatic cells and in germ cells at all times, whereas NHEJ is important in noncycling somatic cells (Clejan et al., 2006).
C. elegans appears to use redundant pathways to cope with DSBs. A genetically distinct pathway involves the ortholog of the human breast cancer 1 gene. BRC-1 in C. elegans was identified using protein-protein interactions (Boulton et al., 2004) and epistatic analysis has shown it to interact with POLQ-1 in a separate pathway than the FA related-(FCD-2 DOG-1)-HR-HEL-308 pathway (Muzzini et al., 2008). Mutants of hel-308 are synthetic lethal with brc-1 mutants, indicating that having one of these pathways functional is essential for survival. Downstream in the process of HR repair, a role for the C. elegans ortholog of the human Holliday junction resolvase GEN-1 has been described both in apoptosis and in DSB repair (Bailly et al., 2010).
PARP-1 (Poly ADP-Ribose polymerase)-1 has a high affinity for SSBs and amongst other functions, recruits factors involved in SSB repair. The PARPs play a major role in regulating replication fork progression and the DDR as well as regulating a number of other biological functions. In humans, PARP inhibitors result in synthetic lethality in BRCA-1 or BRCA-2 defective cells. Although the precise mode of action is unresolved, this synthetic lethal interaction has led to the testing of PARP inhibitors as potential tumor treatments (reviewed by De Vos et al., 2012; Helleday, 2011). PARP and PARG (poly (ADP-ribose) glycohydrolase) orthologs are conserved in C. elegans, making this a model system in which to study their mode of action. In comparison with the 17 components of the PARP family in humans, of which PARP1, PARP2, and PARP3 are known to be involved in DDR (De Vos et al., 2012), there are six orthologs in C. elegans. These, PME 1-6 have been reviewed in St-Laurent and Desnoyers (2011). PME-5 is the ortholog of Tankerase, which is involved in telomere maintenance, spindle assembly and a number of other biological functions. In C. elegans, absence of PARP function is synthetic lethal with cohesion defects, consistent with a role in maintaining genome stability (McLellan et al., 2012).
In order for repair to proceed, chromatid cohesion establishment needs to be coordinated with the repair processes (reviewed in Rudra and Skibbens, 2012; Sjögren and Strom, 2009). The cohesion complex is required for the repair of DSBs by HR so that the broken chromatid can be repaired using an intact sister postreplicatively. Loading of cohesin requires SCC-2. Mutants in both scc-2 and scc-3 are deficient in repair processes such as the apoptotic response, recruitment of the DNA damage sensor 9-1-1 complex, and RAD-51 recombinase response to irradiation-induced DSBs during meiosis (Lightfoot et al., 2011). A temperature-sensitive allele of the cohesin, SMC-3, has been characterized revealing a role for it in DSB repair involving HR repair, even when sister chromatid cohesion was largely intact (Baudrimont, 2011). During meiosis, specialized cohesins are required to ensure proper chromosome segregation (reviewed in Wood et al., 2010). The meiotic specific cohesion, REC-8 and two paralogs, COH-3 and COH-4 (Severson et al., 2009), function along with SMC-5/6 proteins that are required for sister-chromatid-mediated recombination repair (Bickel et al., 2010).
A member of the DOG-1 family of helicases, CHL-1, has been linked to both DNA repair and cohesion (Chung et al., 2011). The chl1 gene in S. cerevisae, when mutated, results in chromosome missegregation and loss (Gerring et al., 1990). The helicase encoded by this gene product has been shown to be required for sister chromatid cohesion (Skibbens, 2004). In humans, a genetic disorder resulting from mutation in the chl-1 gene exhibited aspects of both Fanconi Anemia and cohesinopathy, named Warsaw Breakage Syndrome (van der Lelij et al., 2010). The helicase function of CHL1 has been described (Wu et al., 2011). In C. elegans, chl-1 loss of function results in cell loss both in somatic and germline tissues causing a sterile phenotype. CHL-1 is not required for G-tract stability, but its mutant interaction with DOG-1 mutants provided an assay to examine its role in DNA repair. Mutants in CHL-1 and a mutant in the cohesion core complex, HIM-1/SMC-1, increase deletion formation in a mutant DOG-1 background (Chung et al., 2011) as was shown for mutants in the HR and TLS repair pathways (Youds et al., 2006), linking CHL-1 function to cohesion and FA pathway repair in C. elegans.
Some repair proteins also function in telomere maintenance and their absence generates telomere defects. A classic example in C. elegans is the Mrt (mortality) phenotype, which involves loss of germline immortality. Loss of function of any one of the genes encoding MRTs results in reduced fertility that becomes progressively more severe in subsequent generations until there are no survivors. MRT-2 prevents progressive telomere shortening and accumulation of end-to-end chromosome fusions (Ahmed and Hodgkin, 2000). It encodes a conserved DNA damage checkpoint orthologous to RAD-1 and interacts with other conserved DNA damage checkpoints. The MRT-1 gene product is required for DNA crosslink repair (Meier et al., 2009). MRT-1 has an N-terminal OB-fold domain orthologous to that of POT1 telomere-binding proteins and an exonuclease domain and it acts in the same pathway as telomerase and the 9-1-1 DDR pathways. It is dispensible for DSB repair but functions with the 9-1-1 complex in ICL repair.
Many repair proteins also function in meiosis. Some, such as SPO-11 are meiotic-specific and initiate the crossover event. Others play a role in both somatic and meiotic repair. During meiosis, once the obligate meiotic exchange event has been identified, remaining unrepaired DSBs need to be resolved quickly. Some of the proteins involved in this process are BRC-1 (Adamo et al., 2008) and RTEL-1 (Barber et al., 2008). BRC-1 is the ortholog of BRAC-1, which is associated with breast and ovarian cancers in humans and interacts with FA proteins. In C. elegans BRC-1 has been implicated in sister chromatid repair (Adamo et al., 2008). RTEL-1 is a DOG-1-related helicase that increased crossover frequency in meiosis and was subsequently characterized as an anti-recombinase. First identified in the maintenance of mouse telomere length (Ding et al., 2004), it has been shown to antagonize HR by promoting disassembly of D loop recombination intermediates (Barber et al., 2008). In meiosis, RTEL balances exchange events by promoting exchange between sisters while preventing excess numbers of crossovers between homologs, and has thus been proposed as a component of the molecular basis of crossover interference in C. elegans (Youds et al., 2010). Other repair proteins that have been implicated in the repair of non-crossover DSBs during meiosis, as evidenced by treatment with ionizing radiation, are MUS-81 (Youds et al., 2010), FANCM (Crismani et al., 2012; and see Lorenz and Whitby, 2012), and activated ATM (Couteau and Zetka, 2011). Components of the FA pathway also play meiotic roles, for example, HIM-15/RFS-1 (Yanowitz, 2008). RFS-1 is the sole C. elegans RAD-51 paralogue (Ward et al., 2007) and is proposed to be orthologous to FANCO.
Consequences of damage during cell division in the germ line can be checkpoint-arrest, induced apoptosis, or repair that is either error-prone or error-free. Elimination of cells carrying damaged DNA or faithful repair of the damage will have no consequence for future generations. However checkpoint-bypass or error-prone repair could lead to heritable mutation. Defects in protective proteins such as DOG-1 (Zhao et al., 2008; Cheung et al., 2002), components of the FA pathway such as RFS-1 (Yanowitz, 2008), or checkpoints like ATM (Jones et al., 2012), result in mutator phenotypes. Investigation into the molecular basis of the mutator phenotype of dog-1 mutants resulted in the discovery that DOG-1 is needed to prevent deletions of poly-C/poly-G tracts leading to heritable gene deletions (Cheung et al., 2002) or genome instability due to chromosomal rearrangement (Zhao et al., 2008). In the case of the rfs-1 mutant phenotype, chromosome fusions were accompanied by variations in telomere lengths and sterility (Yanowitz, 2008). Chromosome fusions, reduced viability, and sterility were also observed for atm-1 mutants (Jones et al., 2012). Mutants defective in telomere functions, such as TRT-1 (telomerase reverse transcriptase) or MRT-2 (the DNA damage checkpoint), have progressive loss of telomeric DNA at chromosome ends (Cheung et al., 2006; Meier et al., 2006; Ahmed and Hodgkin, 2000). The shortened telomeres created by loss of MRT function can result in end-to-end chromosome fusions and chromosomal translocations. The types of rearrangements generated in a MRT loss of function background have been analyzed by arrayCGH (comparative genome hybridization) revealing the complex processes that can occur to generate chromosomal instability in the absence of repair (Lowden et al., 2011).
Our understanding of the role of chromatin modifications in the repair process is still in the early stage. Modification and remodeling of the chromatin around the site of damage is known to occur. In the case of DSBs, for example, loss of the heterochromatin associated protein, HP1, results in high sensitivity to both UV light and ionizing radiation, indicating that members of this family of proteins are essential components of DDR systems (Luijsterburg et al., 2009 ). There are three forms of HP1 in mammals and Drosophila, HP1α, β, and γ, which are recruited to DSBs or released from DSBs (Chiolo et al., 2011; Ayoub et al., 2008). Two of these forms are present in C. elegans; removal of one results in hypersensitivity to radiation whereas removal of both results in hypersensitivity to UV-C (Luijsterburg et al., 2009 ).
PARP recruits polycomb components as well as components of the repressive nucleosome remodeling and deacetylase complex to sites of DNA lesions following UV laser microirradiation. Loss of polycomb components results in ionizing radiation (IR) sensitivity. It has been proposed that PARP sets up a transient repressive chromatin structure at sites of DNA damage to block transcription and facilitate repair (Chou et al., 2010).
RNAi depletion of a JMJD2A ortholog resulted in an increase in general H3K9Me3 triggering p53-dependent germline apoptosis (Whetstine et al., 2006). H3K4me2 was affected by SPR-5 activity in the germline, and SPR-5 mutants had increased p53-dependent germ cell apoptosis, increased levels of RAD-51 foci, and sensitivity toward DSB-inducing treatments (Nottke et al., 2011).
At the site of DSBs inactive ATM protein autophosphorylates and dissociates into active kinase monomers. The autophosphorylation is stimulated by acetylation of ATM by histone acetyltransferase and the tumour suppressor protein, Tip60 (Couteau and Zetka, 2011; Fischle, 2009; reviewed in Summers et al., 2011). In C. elegans H2AK5 activation has been observed in the regulation of DSBs on the X-chromosome that become substrate for crossing over and proper disjunction (Wagner et al., 2010). In this study, the regulator of X-chromosome nondisjunction, XND-1, appears to negatively control H2AK5 acetylation allowing the formation of DSBs on the X-chromosome. Also involved is MES-2, the C. elegans ortholog of EZH2 (named for the Drosophila gene, enhancer of zeste, which is a member of the polycomb complex 2). In humans, inherited loss of function of EZH2 results in a rare syndrome including intellectual disability (Gibson et al., 2012), and somatically different modifications of EZH2 are involved in progression to either leukemia or B-cell lymphoma (Morin et al., 2010). The example of EZH2 phenotypes illustrates how much more there is for us to learn in order to understand the role of chromatin modifications in maintaining genome stability.
If the pace of the past few years is any indication, the potential for C. elegans as a model system for the study of DNA repair is very promising. The many aspects of this model system provide opportunities for understanding the interplay between the different pathways and the integration of the many components in different cell types during the stages of development. An exciting new direction is the ability to look comprehensively at the changes involved during repair (or the lack of it) using transcript profiling (Greiss et al., 2008; Harris et al., 2006), RNAseq (Shin et al., 2011) and whole genome sequencing of mutator strains (our lab and others, in progress).
Central to the fidelity of information flow is DNA replication and repair. Previously in WormBook we reviewed research relevant to DNA damage (DNA repair), however there has been an explosion of research in this area over the past six years. C. elegans provides a very powerful model system to investigate comprehensively the integration of repair pathways in a developing animal. Repair in somatic as well as germline cells can be examined, with the germline presenting an exceptional opportunity to examine the progression from mitotically replicating DNA to meiotic tissue to the cycling and eventually differentiating embryo.
A special thanks to David Baillie, Hyeon-Sook Koo, Martin Jones, George Chung, and Ann Marie Howell-Davison for comments and discussion and to Dr. Jeffrey Chu for preparation of Figure 1.
Adamo, A., Collis, S.J., Adelman, C.A., Silva, N., Horejsi, Z., Ward, J.D., Martinez-Perez, E., Boulton, S.J., and LaVolpe, A. (2010). Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol. Cell 39, 25-35. Abstract Article
Adamo, A., Montemauri, P., Silva, N., Ward, J.D., Boulton, S.J., and La Volpe, A. (2008). BRC-1 acts in the inter-sister pathway of meiotic double-strand break repair. EMBO Rep. 9, 287-292. Abstract Article
Ahmed, S., and Hodgkin, J. (2000). MRT-2 checkpoint protein is required for germline immortality and telomere replication in C. elegans. Nature 403, 159-164. Abstract Article
Ameziane, N., Sie, D., Dentro, S., Ariyurek, Y., Kerkhoven, L., Joenje, H., Dorsman, J.C., Ylstra, B. Gille, J.J., Sistermans, E.A., and de Winter, J.P. (2012). Diagnosis of Fanconi anemia: mutation analysis by next-generation sequencing. Anemia 2012, 132856. Abstract Article
Astin, J.W., O'Neil, N.J., and Kuwabara, P.E. (2008). Nucleotide excision repair and t he degradation of RNA pol II by the Caenorhabditis elegans XPA and Rsp5 orthologues, RAD-3 and WWP-1. DNA Repair 7, 267-280. Abstract Article
Ayoub, N., Jeyasekharan, A.D., Bernal, J.A., and Venkitaraman, A.R. (2008). HP1-β mobilization promotes chromatin changes that initiate the DNA damage response. Nature 453, 682-686. Abstract Article
Bailly, A.P., Freeman, A., Hall, J., Declais, A.C., Alpi, A., Lilley, D.M., Ahmed, S., and Gartner, A. (2010). The Caenorhabditis elegans homolog of Gen1/Yen1 resolvases links DNA damage signaling to DNA double-strand break repair. PLoS Genet. 6, e1001025. Abstract Article
Barber, L.J., Youds, J.L., Ward, J.D., McIlwraith, M.J., O'Neil, N.J., Petalcorin, M.I., Martin, J.S., Collis, S.J., Cantor, S.B., Auclair, M., et al. (2008). RTEL-1 maintains genomic stability by suppressing homologous recombination. Cell 135, 261-271. Abstract Article
Baudrimont, A., Penkner, A., Woglar, A., Mamnun, Y.M., Hulek, M., Struck, C., Schnabel, R. Loidl, J., and Jantsch, V. (2011). A new thermosensitive smc-3 allele reveals involvement of cohesin in homologous recombination in C. elegans. PLoS ONE 6, e24799. Abstract Article
Bickel, J.S., Chen, L., Hayward, J., Yeap, S.L., Alkers, A.E., and Chan, R.C. (2010). Structural maintenance of chromosomes (SMC) proteins promote homolog-independent recombination repair in meiosis crucial for germ cell genomic stability. PLoS Genet. 6: e1001028. Abstract Article
Boulton, S.J., Martin, J.S., Polanowska, J., Hill, D.E., Gartner, A., and Vidal, M. (2004). BRCA1/BARD1 orthologs required for DNA repair in Caenorhabditis elegans. Curr. Biol. 14, 33–39. Abstract Article
Bridge, W.L., Vandenberg, C.J., Franklin, R.J. and Hiom, K. (2005). The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nat. Genet. 37, 953-957. Article
Brosh, R.M. (2011). Put on your thinking cap: G-quadruplexes, helicases, and telomeres. Aging 3, 332-335. Abstract
Bunting, S.F., and Nussenzweig, A. (2010). Dangerous liasions: Fanconi anemia and toxic nonhomolougous end joining in DNA crosslink repair. Mol. Cell 39, 164-166. Abstract Article
Cantor, S.B., Bell, D.W., Ganesan, S., Kass, E.M., Drapkin, R., Grossman, S., Wahrer, D.C., Sgroi, D.C., Lane, W.S., Haber, D.A., and Livingston, D.M. (2001). BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 105, 149–160. Abstract Article
Cheung, I., Schertzer, M., Rose, A.M., and Lansdorp, P.M. (2002). Disruption of dog-1 in Caenorhabditis elegans triggers deletions upstream of guanine-rich DNA. Nature Genet. 31, 405–409. Abstract
Cheung, I., Schertzer, M., Rose, A., and Lansdorp, P.M. (2006). High incidence of rapid telomere loss in telomerase-deficient Caenorhabditis elegans. Nucleic Acids Res. 34, 96-103. Abstract Article
Chin G.M. and Villeneuve A.M. (2001). C. elegans mre-11 is required for meiotic recombination and DNA repair but is dispensable for the meiotic G(2) DNA damage checkpoint. Genes Dev. 15, 522-534. Abstract Article
Chiolo, I., Minoda, A., Colmenares, S.U., Polyzos, A., Costes, S.V., and Karpen, G.H. (2011). Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 144, 732-744. Abstract Article
Chou, D.M., Adamson, B., Dephoure, N.E., Tan, X., Nottke, A.C., Hurov, K.E., Gygi, S.P., Colaiacovo, M.P., and Elledge, S.J. (2010). A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. U. S. A. 26, 18475-18480. Abstract Article
Chung, G., O'Neil, N.J., and Rose, A.M. (2011). CHL-1 provides an essential function affecting cell proliferation and chromosome stability in Caenorhabditis elegans. DNA Repair 10, 1174-1182. Abstract Article
Clejan, I., Boerckel, J., and Ahmed, S. (2006). Developmental modulation of nonhomologous end joining in Caenorhabditis elegans. Genetics 173, 1301-1317. Abstract Article
Collis, S.J., Barber, L.J., Ward, J.D., Martin, J.S. and Boulton, S.J. (2006). C. elegans FANCD2 responds to replication stress and functions in interstrand cross-link repair. DNA Repair 5, 1398-1406. Abstract Article
Couteau, F., and Zetka, M. (2011). DNA Damage during meiosis induces chromatin remodeling and synaptonemal complex disassembly. Dev. Cell 20, 353-363. Abstract Article
Crismani, W., Girard, C., Froger, N., Pradillo, M., Santos, J.L., Chelysheva, L., Copenhaver, G.P., Horlow, C., and Mercier, R. (2012). FANCM limits meiotic crossovers. Science 336, 1588-1590. Abstract Article
Deans, A.J., and West, S.C. (2011). DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 11, 467-480. Abstract Article
Denver, D.R., Morris, K., Kewalramani, A., Harris, K.E., Chow, A., Estes, S., Lynch, M., and Thomas, W.K. (2004). Abundance, distribution, and mutation rates of homopolymeric nucleotide runs in the genome of Caenorhabditis elegans. J. Mol. Evol. 58, 584-595. Abstract Article
Derry, W.B., Putzke, A.P., and Rothman, J.H. (2001). Caenorhabditis elegans p53: role in apoptosis, meiosis and stress resistance. Science 294, 591-595. Abstract Article
De Vos, M., Schreiber, V., and Dantzer, F. (2012). The diverse roles and clinical relevance of PARPS in DNA damage repair: current state of the art. Biochem. Pharmacol. 84, 137-146. Abstract Article
Ding, H., Schertzer, M., Wu, X., Gertsenstein, M., Selig, S., Kammori, M., Pourvali, R., Poon, S., Vulto, I., Chavez, E., et al. (2004). Regulation of murine telomere length by Rtel: an essential gene encoding a helicase-like protein. Cell 117, 873-876. Abstract Article
Encalada, S.E., Willis, J., Lyczak, R., and Bowerman, B. (2005). A spindle checkpoint functions during mitosis in the early Caenorhabditis elegans embryo. Mol. Biol. Cell 16, 1056-1070. Abstract Article
Fanconi, G. (1927). Familiare infantile perniziosaartige Anamie (pernizioses Blutbild und Konstitution). Jahrbuch fur Kinderheilkunde und physische Erziehung (Wien) 117, 257-280 (in German) in Lobitz, S. and Velleuer, E. (2006). Guido Fanconi (1892-1979): a jack of all trades. Nat. Rev. Cancer 6, 893-898. Abstract
Fischle, W. (2009). Tip60-ing the balance in DSB repair. Nat. Cell Biol. 11, 1279-1281. Abstract Article
Garcia-Muse, T., and Boulton, S.J. (2005). Distinct modes of ATR activation after replication stress and DNA double-strand breaks in Caenorhabditis elegans. EMBO J. 24, 4345-4355. Abstract Article
Gerring, S.L., Spencer, F., and Hieter, P. (1990). The CHL1 (CTF1) gene product of Saccharomyces cerevisiae is important for chromosome transmission and normal cell cycle progression in G2/M. EMBO J. 9, 4347-4358. Abstract
Gibson, W.T., Hood, R.L., Zhan, S.H., Bulman, D.E., Fejes, A.P., Moore, R., Mungall, A.J., Eydoux, P., Babul-Hirji, R., An, J., et al. (2012). Mutations in EZH2 cause Weaver syndrome. Am. J. Hum. Genet. 90, 110-118. Abstract Article
Greiss, S., Schumacher, B., Grandien, K., Rothblatt, J., and Gartner, A. (2008). Transcriptional profiling in C. elegans suggests DNA damage dependent apoptosis as an ancient function of the p53 family. BMC Genomics 9, 334-344. Abstract Article
Harris, J., Lowden, M., Clejan, I., Tzoneva, M., Thomas, J.H., Hodgkin, J., and Ahmed, S. (2006). Mutator phenotype of Caenorhabditis elegans DNA damage checkpoint mutants. Genetics 174, 601-616. Abstract Article
Helleday, T. (2011). The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 5, 387-393. Abstract Article
Holway, A.H., Kim, S.H., La Volpe, A., and Michael, W.M. (2006). Checkpoint silencing during the DNA damage response in Caenorhabditis elegans embryos. J. Cell Biol. 172, 999-1008. Abstract Article
Hyun, M., Park, S., Kim, E., Kim, D.H., Lee, S.J., Koo, H.S., Seo, Y.S., and Ahn, B. (2012). Physical and functional interactions of Caenorhabditis elegans WRN-1 helicase with RPA-1. Biochemistry 51, 1336-1345. Article
Jaramillo-Lambert, A., Harigaya, Y., Vitt, J. Villeneuve, A., and Engebrecht, J. (2010). Meiotic errors activate checkpoints that improve gamete quality without triggering apoptosis in male germ cells. Curr Biol. 20, 2078-2089. Abstract Article
Jones, M., and Rose, A. (2012). A DOG's view of Fanconi anemia: Insights from C. elegans. Anemia 2012, 323721. Abstract Article
Jones, M.R., Huang, J.C., Chua, S.Y., Baillie, D.L., and Rose, A.M. (2012). The atm-1 gene is required for genome stability in Caenorhabditis elegans. Mol. Genet. Genomics 287, 325-335. Abstract Article
Kim, S.H., and Michael, W.M. (2008). Regulated proteolysis of DNA polymerase η during DNA damage response in C. elegans. Mol. Cell 32, 757-766. Abstract Article
Kitagawa, R., and Rose, A.M. (1999). Components of the spindle-assembly checkpoint are essential in Caenorhabditis elegans. Nat. Cell Biol. 1, 514–521. Abstract Article
Kitao, H., Nanda, I., Sugino, R.P., Kinomura, A., Yamazoe, M., Arakawa, H., Schmid, M., Innan, H., Hiom, K., and Takata, M. (2011). FancJ/Brip1 helicase protects against genomic losses and gains in vertebrate cells. Genes Cells 16, 714-727. Abstract Article
Ko, E., Lee, J., and Lee, H. (2008). Essential role of brc-2 in chromosome integrity of germ cells in C. elegans. Mol. Cells 26, 590-594. Abstract
Kottemann, M.C,. and Smogorzewska, A. (2013). Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 493, 356-363. Abstract Article
Kratz, K., Schopf, B., Kaden, S., Sendoel, A., Eberhard, R., Lademann, C., Cannavo, E., Sartori, A., Hengartner, M.O., and Jiricny, J. (2010). Deficiency of FANCD2-associated nuclease KIAA1018/FAN1 sensitizes cells to interstrand crosslinking agents. Cell, 142, 77-88. Abstract Article
Kruisselbrink, E., Guryev, V., Brouwer, K., Pontier, D.B., Cuppen, E., and Tijsterman, M. (2008). Mutagenic capacity of endogenous G4 DNA underlies genome instability in FANCJ-defective cells in C. elegans. Curr. Biol. 18, 900-905. Abstract Article
Lange, J., Pan, J., Cole, F., Thelen, M.P., Jasin, M., and Keeney, S. (2011). ATM controls meiotic double-strand-break formation. Nature 479, 237-240. Article
Lans, H., and Vermeulen, W. (2011). Nucleotide excision repair in Caenorhabditis elegans. Mol. Biol. Int. 2011, 542795. Abstract Article
Lans, H., Marteijn, J.A., Schumacher, B., Hoeijmakers, J.H.J., Jansen, G., and Vermeulen, W. (2010). Involvement of global genome repair, transcription coupled repair and chromatin remodeling in UV DNA damage response changes during development. PLoS Genet. 6, e1000941. Abstract Article
Lee, K.Y., Chung, K.Y., and Koo, H.S. (2010). The involvement of FANCM, FANCI and checkpoint proteins in the interstrand DNA crosslink repair pathway is conserved in C. elegans. DNA Repair 9, 374-382. Abstract Article
Lee, K.Y., Yang, I., Park, J.E., Baek, O.R., Chung, K.Y., and Koo, H.S. (2007). Developmental stage and DNA damage-specific functions of C. elegans FANCD2. Biochem. Biophys. Res. Commun. 352, 479-485. Abstract Article
Lee, M.H., Hollis, S.E., Yoo, B.H., and Nykamp, K. (2011). Caenorhabditis elegans DNA-2 helicase/endonuclease plays a vital role in maintaining genome stability, morphogenesis, and life span. Biochem. Biophys. Res. Commun. 407, 495-500. Article
Lee, S.J., Gartner, A., Hyun, M., Ahn, B., and Koo, H.S. (2010). The Caenorhabditis elegans Werner syndrome protein functions upstream of ATR and ATM in response to DNA replication inhibition and double-strand DNA breaks. PLoS Genet. 6, e1000801. Abstract Article
Lemmens, B.B., and Tijsterman, M. (2011). DNA double-strand break repair in Caenorhabditis elegans. Chromosoma 120, 1-21. Abstract Article
Li, R., and Murray, A.W. (1991). Feedback control of mitosis in budding yeast. Cell 66, 519–531. Abstract Article
Lightfoot, J., Testori, S., Barroso, C., and Martinez-Perez, E., (2011). Loading meiotic cohesion by SCC-2 is required for early processing of DSBs and for the DNA damage checkpoint. Curr Biol. 21, 1421-1430. Abstract Article
Liu, T., Ghosal, G., Yuan, J., Chen, J., and Huang, J. (2010). FAN1 acts with FANCI-FAND2 to promote DNA interstrand cross-link repair. Science 329, 693-696. Abstract Article
Lobitz, S., and Velleuer, E. (2006). Guido Fanconi (1892-1979): a jack of all trades. Net. Rev. Cancer 6, 893-898. Abstract Article
Lorenz, A., and Whitby, M.C. (2012). How not to get cross(ed): a novel role for FANCM in meiotic recombination. Cell Cycle 11, 3347-3348. Abstract Article
Lowden, M.R., Flibotte, S., Moerman, D.G., and Ahmed, S. (2011). DNA synthesis generates terminal duplications that seal end-to-end chromosome fusions. Science 332, 468-471. Abstract Article
Luijsterburg, M.S., Dinant, C., Lans, H., Stap, J., Wiernasz, E., Lagerwerf, S., Warmerdam, D.O., Lindh, M., Brink, M.C., Dobrucki, J.W., et al. (2008). Heterochromatin protein 1 is recruited to various types of DNA damage. J. Cell Biol. 185, 577-586. Article
Maizels, N. (2008). Genomic stability: FANCJ-dependent G4 DNA repair. Curr. Biol. 18, R613-R614. Abstract Article
Martin, J.S., Winkelmann, N., Petalcorin, M.I., McIlwraith, M.J., and Boulton, S.J. (2005). RAD-51-dependent and –independent roles of a Caenorhabditis elegans BRCA2-related protein during DNA double-strand break repair. Mol. Cell. Biol. 25, 3127-3139. Abstract Article
Mackay, C., Declais, AC., Lundin, C., Agostinho, A., Deans, A.J., Macartney, T.J., Hofmann, K., Gartner, A., West, S.C., Helleday, T., et al. (2010). Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell 142, pp. 65-76. Abstract Article
McLellan, J.L., O'Neil, N.J., Barrett, I., Ferree, E., van Pel, D.M., Ushey, K., Sipahimalani, P., Bryan, J., Rose, A.M.. and Hieter, P. (2012). Synthetic lethality of cohesions with PARPs and replication fork mediators. PLoS Genet. 8, e1002574. Abstract Article
McLellan, J., O'Neil, N. Tarailo, S., Stoepel, J., Bryan, J., Rose, A., and Hieter, P. (2009). Synthetic lethal genetic interactions that decrease somatic cell proliferation in Caenorhabditis elegans identify the alternative RFC CTF18 as a candidate cancer drug target. Mol. Biol. Cell 20, 5306-5313. Abstract Article
McVey, M. (2010). Strategies for DNA interstrand crosslink repair: insights from worms, flies, frogs, and slime molds. Environ. Mol. Mutagen. 51, 646-658. Abstract
Meier, B., Barber, L.J., Liu, Y., Shtessel, L., Boulton, S.J., Gartner, A., and Ahmed, S. (2009). The MRT-1 nuclease is required for DNA crosslink repair and telomerase activity in vivo in Caenorhabditis elegans. EMBO J. 28, 3549-3563. Abstract Article
Meier, B., Clejan, I., Liu, Y., Lowden, M., Gartner, A., Hodgkin, J., and Ahmed, S. (2006). trt-1 is the Caenorhabditis elegans catalytic subunit of telomerase. PLoS Genet. 2, e18. Abstract Article
Morin, R.D., Johnson, N.A., Severson, T.M., Mungall, A.J., An, J., Goya, R., Paul, J.E., Boyle, M., Woolcock, B.W., Kuchenbauer, F., et al. (2010). Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 42, 181-185. Abstract Article
Muzzini, D.M., Plevani, P., Boulton, S.J., Cassata, G., and Marini, F. (2008). Caenorhabditis elegans POLQ-1 and HEL-308 function in two distinct DNA interstrand cross-link pathways. DNA Repair 7, 941-950. Abstract Article
Nottke, A.C., Beese-Sims, S.E., Pantalena, L.F., Reinke, V., Shi, Y., and Colaiacovo, M.P. (2011). SPR-5 is a histone demethylase with a role in meiotic double-strand break repair. Proc. Natl. Acad. Sci. U. S. A. 108, 12805-12810. Abstract Article
O'Neil, N. and Rose, A. DNA repair (January 13, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.54.1, http://www.wormbook.org.
Pace, P., Mosedale, G., Hodskinson, M., Rosado, I.V., Sivasubramaniam, M., and Patel, K.J. (2010). Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science 329, 219-223. Abstract Article
Petalcorin, M.I., Galkin, V.E., Yu, X., Egelman, E.H., and Boulton, S.J. (2007). Stabilization of RAD-51-DNA filaments via an interaction domain in Caenorhabditis elegans BRCA2. Proc. Natl. Acad. Sci. U. S. A. 104, 8299-82304. Abstract Article
Pontier, D.B., and Tijsterman, M. (2009). A robust network of double-strand break repair pathways governs genome integrity during C. elegans development. Curr. Biol. 19, 1384-1388. Abstract Article
Rodriguez R., Miller K.M., Forment J.V., Bradshaw C.R., Nikan M., Britton S., Oelschlaegel T., Xhemalce B., Balasubramanian S., and Jackson S.P. (2012). Small-molecule-induced DNA damage identifies alternative DNA structures in human genes. Nat. Chem. Biol. 8, 301-310. Abstract Article
Roerink, S.F., Koole, W., Stapel, L.C., Romeijn, R.J., and Tijsterman, M. (2012) A broad requirement for TLS polymerases η and κ, and interacting sumoylation and nuclear pore proteins, in lesion bypass during C. elegans embryogenesis. PLoS Genet. 8, e1002800. Abstract Article
Rudra, S. Skibbens, R.V. (2012). Sister chromatid cohesion establishment occurs in concert with lagging strand synthesis. Cell Cycle 11, 2114-2121. Abstract Article
Saito, T.T., Youds, J.L., Boulton, S.J., and Colaiácovo, M.P. (2009). Caenorhabditis elegans HIM-18/SLX-4 interacts with SLX-1 and XPF-1 and maintains genomic integrity in the germline by processing recombination intermediates. PLoS Genet. 5, e1000735. Abstract Article
Schumacher, B., Hofmann, K., Boulton, S., and Gartner, A. (2001). The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Curr. Biol. 11, 1722-1727. Abstract Article
Sen, D., and Gilbert, W. (1988). Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature 334, 364-366. Abstract Article
Severson, A.F., Ling, L., van Zuylen, V., and Meyer, B.J. (2009). The axial element protein HTP-3 promotes cohesion loading and meiotic axis assembly in C. elegans to implement the meiotic program of chromosome segregation. Genes Dev. 23, 1763-1778. Abstract Article
Shin, H., Lee, H., Fejes, A.P., Baillie, D.L., Koo, H.S., and Jones, S.J. (2011). Gene expression profiling of oxidative stress response of C. elegans aging defective AMPK mutants using massively parallel transcriptome sequencing. BMC Res. Notes 8, 34. Abstract Article
Skibbens, R.V. (2004). Chl1p, a DNA helicase-like protein in budding yeast, functions in sister-chromatid cohesion. Genetics 166, 33-42. Abstract Article
Smogorzewska, A., Desetty, R., Saito, T.T., Schlabach, M., Lach, F.P., Sowa, M.E., Clark, A.B., Kunkel, T.A., Harper, J.W., Colaiacovo, M.P., and Elledge, S.J. (2010). A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol. Cell 39, 36-47. Abstract Article
Sjögren, C., and Ström, L. (2009). S-phase and DNA damage activated establishment of sister chromatid cohesion–importance for DNA repair. Exp. Cell Res. 316, 1445-1453. Abstract Article
Stergiou, L. Doukoumetzidis, K., Sendoel, A., and Hengartner, M.O. (2007). The nucleotide excision repair pathway is required for UV-C-induced apoptosis in Caenorhabditis elegans. Cell Death Differ. 14, 1129-1138. Abstract Article
Stergiou, L. Eberhard, R., Doukoumetzidis, K., and Hengartner, M.O. (2011). NER and HR pathways act sequentially to promote UV-C-induced germ cell apoptosis in Caenorhabditis elegans. Cell Death Differ. 18, 897-906. Abstract Article
St-Laurent, J-F., and Desnoyers, S. (2011). Poly(ADP-Ribose) metabolism analysis in the nematode Caenorhabditis elegans. Methods Mol. Biol. 780, 413-425. Abstract Article
Summers, K.C., Shen, F., Potchanant, S., Phipps, E.A., Hickey, R.J., and Malkas, L.H. (2011). Phosphorylation: the molecular switch of double-stand break repair. Int. J. Proteomics 2011, 373816. Abstract Article
Takata, M., Yamamoto, K., Matsushita, N., Kitao, H., Hirano, S., and Ishiai, M. (2006). The Fanconi anemia pathway promotes homologous recombination repair in DT40 cell line. Subcell. Biochem. 40, 295-311. Abstract
Van der Lelij, P., Chrzanowska, K.H., Godthelp, B.C., Rooimans, M.A., Oostra, A.B., Stumm, M., Zdzienicka, M.X., Joenje, H., and de Winter, J.P. (2010). Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am. J. Hum. Genet. 86, 262-266. Abstract Article
Vermezovic, J., Stergiou, L., Hengartner, M.O., and d'Adda di Fagagna, F., (2012). Differential regulation of DNA damage response activation between somatic and germline cells in Caenorhabditis elegans. Cell Death Differ. 19, 1847-1855. Abstract
Wagner, C.R., Kuervers, L., Baillie, D.L., and Yanowitz, J.L. (2010). xnd-1 regulates the global recombination landscape in Caenorhabditis elegans. Nature 467, 839-843. Abstract Article
Ward, J.D., Barber, L.J., Petalcorin, M.I., Yanowitz, J., and Boulton, S.J. (2007). Replication blocking lesions present a unique substrate for homologous recombination. EMBO J. 26, 3384-3396. Abstract Article
Watrin, E., and Peters, J.M. (2006). Cohesin and DNA damage repair. Expe. Cell Res. 312, 2687-2693. Abstract Article
Weidhaas, J.B., Eisenmann, D.M., Holub, J.M., and Nallur, S.V. (2006). A Caenorhabditis elegans tissue model of radiation-induced reproductive cell death. Proc. Natl. Acad. Sci. U. S. A. 103, 9946-9951. Abstract Article
Whetstine, J.R., Nottke, A., Lan, F., Huarte, M., Smolikov, S., Chen, Z., Spooner, E., Li, E., Zhang, G., Colaiacovo, M., and Shi, Y. (2006). Reversal of histone lysine trimethylation by JMJD2 family of histone demethylases. Cell 125, 467-481. Abstract Article
Wood, A.J., Severson, A.F., and Meyer, B.J. (2010). Condensin and cohesion complexity: the expanding repertoire of functions. Nat. Rev. Genet. 11, 391-404. Abstract Article
Wu, Y., and Brosh, R.M., Jr. (2010). G-quadruplex nucleic acids and human disease. FEBS J. 277, 3470-3488. Abstract Article
Wu, Y., Sommers, J.A., Khan, I., de Winter, J.P., and Brosh, R.M., Jr. (2011). Biochemical characterization of Warsaw breakage syndrome helicase. J. Biol. Chem. 287, 1007-1021. Abstract Article
Yanowitz, J.L. (2008). Genome integrity is regulated by the Caenorhabditis elegans Rad51D homolog rfs-1. Genetics 179, 249-262. Abstract Article
Youds, J.L., Barber, L.J., and Boulton, S.J. (2009). C. elegans: a model of Fanconi anemia and ICL repair. Mutat. Res. 668,103-116. Abstract Article
Youds, J.L., Barber, L.J., Ward, J.D., Collis, S.J., O'Neil, N.J., Boulton, S.J., and Rose, A.M. (2008). DOG-1 is the Caenorhabditis elegans BRIP1/FANCJ homologue and functions in interstrand cross-link repair. Mol. Cell. Biol. 28, 1470-1479. Abstract Article
Youds, J.L., O'Neil, N.J., and Rose, A.M. (2006). Homologous recombination is required for genome stability in the absence of DOG-1 in Caenorhabditis elegans. Genetics 173, 697-708. Abstract Article
Youds, J.L., Mets, D.G., McIlwraith, M.J., Martin, J.S., Ward, J.D. O'Neil, N.J., Rose, A.M., West, S.C., Meyer, B.J., and Boulton, S.J. (2010). RTEL-1 enforces meiotic crossover interference and homeostasis. Science 327, 1254-1258. Abstract Article
*Edited by Thomas Blumenthal, Last revised: February 14, 2013. Published December 4, 2014. This chapter should be cited as: Rose A. Replication and repair (December 4, 2014), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.54.2, http://www.wormbook.org.
Copyright: © 2014 Ann Rose. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed: [email protected]
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.