Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
Neurotransmitters signal via G protein coupled receptors (GPCRs) to modulate activity of neurons and muscles. C. elegans has ∼150 G protein coupled neuropeptide receptor homologs and 28 additional GPCRs for small-molecule neurotransmitters. Genetic studies in C. elegans demonstrate that neurotransmitters diffuse far from their release sites to activate GPCRs on distant cells. Individual receptor types are expressed on limited numbers of cells and thus can provide very specific regulation of an individual neural circuit and behavior. G protein coupled neurotransmitter receptors signal principally via the three types of heterotrimeric G proteins defined by the G alpha subunits Gαo, Gαq, and Gαs. Each of these G alpha proteins is found in all neurons plus some muscles. Gαo and Gαq signaling inhibit and activate neurotransmitter release, respectively. Gαs signaling, like Gαq signaling, promotes neurotransmitter release. Many details of the signaling mechanisms downstream of Gαq and Gαs have been delineated and are consistent with those of their mammalian orthologs. The details of the signaling mechanism downstream of Gαo remain a mystery. Forward genetic screens in C. elegans have identified new molecular components of neural G protein signaling mechanisms, including Regulators of G protein Signaling (RGS proteins) that inhibit signaling, a new Gαq effector (the Trio RhoGEF domain), and the RIC-8 protein that is required for neuronal Gα signaling. A model is presented in which G proteins sum up the variety of neuromodulator signals that impinge on a neuron to calculate its appropriate output level.
The nervous system functions through the use of neurotransmitters that act as chemical signals between cells. Small-molecule neurotransmitters such as acetylcholine, glutamate, and GABA (gamma aminobutyric acid) are released from vesicles clustered at synapses. Neuropeptides (secreted proteins of <50 amino acids) are released at synaptic and non-synaptic sites from a different class of vesicles, known as dense-core vesicles for their appearance in the electron microscope (Merighi et al., 2011). Certain small-molecule neurotransmitters such as serotonin can be released from either class of vesicle. All these types of neurotransmitters act on neurons and muscles to generate dynamic patterns of activity that constitute thoughts and behaviors. A principal objective in neuroscience is to understand the molecular mechanisms by which neurons and muscles respond to neurotransmitters. This objective is important to advance our basic science understanding of the brain, but also is of great medical significance since many pharmaceuticals used to treat psychiatric disorders act by mimicking, antagonizing, or altering the levels of naturally-occurring neurotransmitters (Conn and Roth, 2008), and drugs of abuse also act by altering neurotransmitter signaling (Joffe et al., 2014).
Neurotransmitters signal via two distinct classes of receptors, known within the neuroscience field as ionotropic and metabotropic receptors. Ionotropic receptors are neurotransmitter-gated ion channels, and most small-molecule neurotransmitters each have a number of such receptors (Lemoine et al., 2012). Binding of neurotransmitter to an ionotropic receptor favors channel opening, and communication between neurons using ionotropic receptors can occur in less than a millisecond (Sabatini and Regehr, 1996). Metabotropic receptors are known outside the neuroscience field as G protein coupled receptors (GPCRs) because they activate intracellular signaling proteins called heterotrimeric G proteins. All small-molecule neurotransmitters have G protein coupled receptors, as do most neuropeptides (Hall, 2004; Beaulieu and Gainetdinov, 2011; Pytliak et al., 2011; Hoyer and Bartfai, 2012; Vaidya et al., 2013; Kruse et al., 2014). An individual small-molecule neurotransmitter might have up to a dozen different GPCRs. There are over 100 neuropeptide genes in both humans and C. elegans, and each organism also has about an equal number of GPCRs that are likely to be neuropeptide receptors, as they are similar to the few G protein coupled neuropeptide receptors that have been characterized so far (Li and Kim, 2008; Li and Kim, 2010; Frooninckx et al., 2012; Hoyer and Bartfai, 2012). Binding of a neurotransmitter to a GPCR, as opposed to an ionotropic receptor, leads to slower and longer lasting effects. For example, GCPR signaling can involve biochemical amplification of a signal (e.g., production of a pool of second messenger) that is much slower than the rapid voltage changes induced by opening ion channels, and GPCR signaling can result in changes to the transcriptional program and structure of a neuron that last days or longer (Kandel, 2004). Whereas ionotropic receptors mediate signaling underlying such prosaic neural functions as the knee-jerk response, GPCRs mediate signaling underlying more poetical functions of the brain, such as feelings of pleasure and love that result from dopamine and oxytocin (Love, 2014), the psychedelic effects of hallucinogens (Fantegrossi et al., 2008), and the regulation of mood by serotonin (Donaldson et al., 2013).
This review focuses on insights into the molecular mechanisms and biological functions of neurotransmitter signaling through GPCRs that arise from studies in the model organism C. elegans. But why study neurotransmitter signaling in the worm? Signaling through ionotropic receptors has already been studied in a sophisticated manner in other species using electrophysiological techniques to analyze ion channel activity. Such electrophysiological studies have often used model organisms such as slug, squid, leech, or crab that have large neurons easily accessible to electrodes. GPCRs activate intracellular signaling pathways that can have indirect effects on ion channel activity, and electrophysiological studies in the same model organisms have given important insights into how GPCR signaling modulates the function of neural circuits (Bailey and Kandel, 2008; Marder, 2012). However, methods for studying intracellular signaling pathways have been most highly developed outside of the neuroscience field, with the greatest successes coming from applying a combination of biochemistry and genetics, techniques not easily applied in the model organisms suited to electrophysiology. A remarkable body of biochemical studies of signaling by heterotrimeric G proteins, mostly in non-neuronal mammalian cells, has been in progress for decades and has resulted in many Nobel prizes (Cori and Cori, 1947; Sutherland, 1971; Krebs, 1992; Fischer 1992; Gilman 1994; Rodbell 1994; Kobilka, 2013; Lefkowitz 2013). Prior to the studies in C. elegans described in this review, neural G protein signaling had not been seriously studied using genetic approaches. Thus, despite the excellence of the body of biochemical work on G protein signaling, there remained significant gaps in understanding the molecular mechanisms of this type of signaling in neurons, and also in achieving a big-picture understanding of how and why such signaling is used to control the activity of neural circuits.
As described in this review, the molecular mechanisms of neural G protein signaling are strongly conserved between humans and C. elegans, and C. elegans provides two advantages that complement the past electrophysiological and biochemical work on neural G protein signaling. First, the power of forward genetics in C. elegans has allowed new neural signaling proteins to be discovered. Second, the simplicity of the C. elegans nervous system, combined with the use of genetics, has allowed biological functions to be assigned to signaling by specific neurotransmitters acting through specific GPCRs on individual identified neurons. Enough such biological functions of neural G protein signaling have now been described that the long-elusive big-picture understanding of the whole purpose of this mode of neurotransmission is beginning to emerge.
Here I outline the activity cycle for a generic heterotrimeric G protein as a prelude to diving more deeply into how specific GPCRs, G proteins, and their downstream signaling pathways regulate neural function in C. elegans. The G protein activity cycle proceeds through five states labeled in Figure 1. Most steps in the cycle were discovered and characterized through biochemical studies of mammalian proteins, but the RGS and RIC-8 proteins that catalyze the transitions between states 4 and 5 were discovered through studies in C. elegans and subsequently characterized in mammals.
|
Figure 1. Activity cycle for a generic heterotrimeric G protein. In the inactive state (1), the G protein is an αβγ heterotrimer bound to GDP. When neurotransmitter binds the receptor (2), it assumes an active conformation that can bind the G protein heterotrimer and induce a conformational change in Gα that releases GDP. GTP can then bind the open nucleotide site (3), inducing another conformational change in Gα that releases both the receptor and Gβγ. The separate Gα-GTP and Gβγ complexes can then associate with their respective effector proteins (4), which are typically transmembrane protein complexes, activating those effectors for downstream signaling. Gα signaling is terminated when a Regulator of G protein Signaling (RGS) protein induces Gα to hydrolyze GTP (5), and Gβγ signaling is terminated when Gβγ re-associates with Gα-GDP (1). The RIC-8 non-receptor nucleotide-exchange protein, working with GPR domain proteins, can prolong signaling by converting Gα-GDP (5) back to Gα-GTP (4). RIC-8 is also a chaperone required for Gα folding and stability, which may be its principal function.
GPCRs are integral membrane proteins with seven transmembrane helices. Heterotrimeric G proteins have α, β, and γ subunits and are peripheral membrane proteins tethered to the inner leaflet of the plasma membrane by lipid modifications (Hepler and Gilman, 1992; Wedegaertner et al., 1995). The Gα subunit changes conformation depending on whether it is bound to GDP, GTP, or no guanine nucleotide, and these conformational changes allow Gα to alter its association with other proteins (Noel et al., 1993; Coleman et al., 1994; Lambright et al., 1994; Wall et al., 1995). In its GDP-bound inactive state, Gα is typically bound to Gβγ to form an inactive G protein heterotrimer.
Neurotransmitter binding induces conformational shifts in GPCRs that have recently been delineated by X-ray crystallographic studies (Katritch et al., 2013). GPCRs and G proteins are able to diffuse laterally due to the fluid nature of biological membranes, and a neurotransmitter-bound, activated receptor can thus collide with an inactive G protein heterotrimer. This interaction induces a large conformational change in Gα that allows the release of bound GDP (Fung et al., 1981; Brandt and Ross, 1986; Rasmussen et al., 2011). The active GPCR and nucleotide-free G protein heterotrimer form a stable complex that dissociates in the next step of the G protein cycle.
The open nucleotide-binding site on Gα can bind GTP, which causes Gα to undergo another conformational change that causes Gα-GTP to dissociate from both the receptor and Gβγ (Noel et al., 1993; Coleman et al., 1994; Lambright et al., 1994; Wall et al., 1995). The release of GDP and subsequent GTP binding by Gα is referred to as “nucleotide exchange”. The receptor, which remains bound to neurotransmitter, can diffuse laterally in the membrane and proceed to promote nucleotide exchange on additional G proteins. Thus the active receptor can function as an enzyme to catalytically promote conversion of inactive Gαβγ-GDP to the active signaling species Gα-GTP and Gβγ (Ross, 2014).
The separated Gα-GTP and Gβγ complexes are able to stably bind to and regulate activity of other proteins to promote responses in the cell, and these other proteins are collectively termed G protein “effectors”. Effectors have been identified for Gβγ (Reuveny et al., 1994; Herlitze et al., 1996) and for the Gα isoforms Gαs (Sunahara et al., 1996), Gαq (Kadamur and Ross, 2013; Rohas et al., 2007; Williams et al., 2007), Gα12 (Siehler, 2009), and Gαi (Taussig et al., 1994). Most G protein effectors are transmembrane protein complexes. Some G protein effectors are enzymes that catalyze the production of second messengers, small molecules that can diffuse in the cell and evoke responses. Gα proteins bind their effectors via the same “switch” regions they use to bind Gβγ. Gα must be in its GTP-bound conformation to bind and activate effectors, but must be in its GDP-bound conformation to bind Gβγ. Thus Gα serves as a molecular switch that alternates between an active GTP-bound state and an inactive GDP-bound state. Gβγ can only activate its effectors once it has dissociated from Gα.
Gα subunits of heterotrimeric G proteins have a slow but significant intrinsic GTPase activity, such that purified Gα protein can hydrolyze bound GTP to GDP with a half time on the order of a few minutes. A key contribution of C. elegans and yeast genetics was the discovery of a class of “regulator of G protein signaling” (RGS) proteins (Koelle and Horvitz, 1996; Dohlman et al., 1996) that serve as GTPase activators for Gα proteins, speeding up the hydrolysis reaction by orders of magnitude (Berman et al., 1996; Tesmer et al., 1997). Physiologically, Gα signaling is typically inactivated with the help of an RGS protein (Figure 1).
A second key contribution of C. elegans genetics was the discovery of RIC-8, a soluble protein that promotes G protein signaling (Miller et al., 2000), sometimes with the help of other proteins containing the G protein regulatory (GPR) motif that binds Gα proteins (Colombo et al., 2003; Gotta et al., 2003; Srinivasan et al., 2003; Hofler and Koelle, 2011). Genetic studies in C. elegans show that GPR proteins and RIC-8 promote G protein activity, and the biochemical activities of these proteins in vitro suggest that they may do so by reactivating Gα-GDP as illustrated in Figure 1. However, studies in mammalian cells show that RIC-8, in addition to catalyzing nucleotide exchange in vitro, also acts in living cells as a chaperone to promote folding and stability of Gα proteins (Gabay et al., 2011; Chan P. et al., 2013). The C. elegans genetic data are consistent with the alternative model that GPR and RIC-8 proteins are simply required for Gα folding and stability.
The G protein activity cycle is completed when Gα-GDP re-associates with Gβγ to re-form the inactive Gαβγ heterotrimer. Gβγ is sequestered in the heterotrimer so that it can no longer associate with its effectors.
In this section, I describe analysis of the surprisingly large set of neurotransmitters and neural G protein coupled receptors present in C. elegans. Each neurotransmitter and each receptor is expressed in a very specific and limited set of neurons, and as a result each affects a very specific and limited set of behaviors. An important result from C. elegans is that a neurotransmitter can signal via a GPCR expressed on cells that are not postsynaptic to the neurons that release that neurotransmitter. Thus neurotransmitters travel through tissue to signal at sites distant from their site of release. The pattern of neurotransmitter signaling via GPCRs is thus not determined by the synaptic wiring of the nervous system, but rather by the specific expression patterns of the neurotransmitters and their GPCR receptors. The widespread nature of extrasynaptic neurotransmitter signaling forces us to expand our notion of a neural circuit to a unit consisting of neurons that function together but that may lack direct anatomical connections.
Most major small-molecule neurotransmitters used in humans are also found in C. elegans (exceptions are norepinephrine, histamine, and possibly glycine and ATP). Table 1 and Table 2 summarize studies of a set of 28 C. elegans GPCRs for small-molecule neurotransmitters. This includes four serotonin receptors, four dopamine receptors, three octopamine receptors, three tyramine receptors, three acetylcholine receptors, two GABA receptors that function together as an obligate dimer, three apparent glutamate receptors, and six additional predicted receptors with sequence similarity to small molecule neurotransmitter GPCRs that have yet to have ligands assigned to them or to be studied genetically. Many of these 28 GPCRs are produced in multiple isoforms via alternative splicing of their RNA transcripts
Putative small-molecule neurotransmitter GPCRs were first identified in C. elegans as homologs of mammalian neurotransmitter receptors (e.g., Komuniecki et al., 2004). Such analyses can make predictions for the ligand that might activate a GPCR homolog, but these are weak predictions. For example, the C. elegans receptors SER-2, SER-3, and SER-6 were originally assigned their names due to similarity with serotonin receptors, but later proved experimentally to be receptors for other biogenic amine neurotransmitters (Table 1). Thus, assigning a ligand to a GPCR requires experimental evidence. Three lines of such experimental evidence are described below, and Table 1 lists which of these lines of evidence are available for each of 22 C. elegans receptors.
The first line of experimental evidence is listed in Table 1 as “binding studies”. Here, a GPCR is expressed in heterologous cells inducing a binding activity in their membranes for a radiolabeled ligand. Since characterizing binding by many different radioligands is inconvenient, typically a single radioligand with binding activity is identified (e.g., 3H-LSD, which binds with high affinity to most biogenic amine receptors), and then the ability of many unlabeled neurotransmitters and pharmaceuticals to compete off the radioligand is measured as a “Ki”. Such studies measure the relative binding affinity of different ligands, but do not determine if an individual ligand is an agonist (activator) or antagonist (inhibitor) for the receptor. Based on such binding data, a GPCR is considered likely to be a receptor for the neurotransmitter that appears to bind it with highest affinity.
The second line of experimental evidence listed in Table 1 is “heterologous cell signaling”. Here, a GPCR is expressed in heterologous cultured cells, typically mammalian cells or Xenopus oocytes, potential ligands are applied in the medium, and activation of downstream signaling is measured (e.g., through use of fluorescent Ca2+ indicators, electrophysiological recording of G protein regulated ion channels, etc.). This method allows the concentration of a ligand that gives half-maximal response (EC50) to be measured, and can determine if a ligand is an agonist or antagonist. Further, it provides evidence for the type of Gα protein activated by the receptor (see Section 2.2.2.). Based on such heterologous cell signaling studies, a GPCR is considered likely to be a receptor for the neurotransmitter that activates it with the lowest EC50.
The third line of experimental evidence listed in Table 1 is genetic studies in C. elegans. In many cases, a mutant for a GPCR renders worms insensitive to the effects of a specific neurotransmitter, either applied exogenously or released endogenously via optogenetic stimulation. Another type of genetic study demonstrates that a GPCR mutant shows behavioral defects similar to those of a mutant lacking a particular neurotransmitter. Such genetic studies provide evidence that GPCR functions in vivo as a receptor for a specific neurotransmitter.
Table 1 shows that for the 22 GPCRs listed as assigned a ligand, the number of experimental lines of evidence for that assignment varies from zero to three, and the quality of each piece of evidence can also vary widely. Clearly, the more experimental evidence that is available, the more confident a ligand assignment will be.
Both C. elegans and mammals have multiple GPCRs for individual neurotransmitters. A large number of pharmaceuticals have been developed that activate or antagonize specific mammalian receptor isoforms. It is tempting to try to use C. elegans as a model system to investigate the functions of specific neurotransmitter receptor isoforms to better understand the actions of these important drugs. However, when the sequences of C. elegans neurotransmitter receptors are lined up with those of mammalian receptors, individual C. elegans receptors cannot be unambiguously assigned as orthologs of specific mammalian receptors. For example, the C. elegans serotonin receptors are more similar to mammalian serotonin receptors than they are to mammalian receptors for other neurotransmitters, but an additional C. elegans receptor (named SER-2) is also most similar to mammalian serotonin receptors in sequence and turned out be a tyramine receptor (Rex et al., 2004). By sequence analysis, the bona fide C. elegans serotonin receptors do not unambiguously match up with individual mammalian serotonin receptor isoforms. Furthermore, when C. elegans serotonin receptors are expressed in cultured cells or Xenopus oocytes, their profiles of binding and activation by drugs do not match up to the pharmacology of specific mammalian serotonin receptor isoforms (Komuniecki et al., 2004). Thus, while C. elegans generally provides an excellent model for studying small-molecule neurotransmitter signaling through G proteins, there are limits to the level of conservation of receptors between humans and worms.
An important issue in assigning ligands to GPCRs is the potential that a single GPCR might physiologically mediate signaling by more than one type of neurotransmitter. The heterologous cell signaling studies cited in Table 1 show that certain receptors have significant affinities for more than one biogenic amine neurotransmitter. For example, the dopamine receptor DOP-3 is activated in heterologous cell signaling studies by dopamine with an EC50 of 27 nm, but also by tyramine with an EC50 of 500 nm (Sugiura et al., 2005). Binding studies show significant affinities for more than one biogenic amine by the receptors DOP-1, DOP-2 (Suo et al., 2002), OCTR-1 (Wragg et al., 2007), SER-2 (Rex and Komuniecki, 2002), TYRA-2 (Rex et al., 2005), and TYRA-3 (Wragg et al., 2007). Since the synaptic concentrations of released neurotransmitters are thought to go above one millimolar (Barberis et al., 2011), the high nanomolar or micromolar binding affinities of certain receptors for a secondary neurotransmitter could be biologically meaningful. Studies in C. elegans could resolve this issue. Given the complete wiring diagram in C. elegans, along with the known identifies of neurons that release specific neurotransmitters and that express specific GPCRs, there is the potential to identify synapses at which a GPCR is exposed to a high concentration of one of its lower-affinity ligands. Genetic analysis could then determine if this results in physiologically significant signaling.
Sequence comparison of C. elegans GPCRs to homologous mammalian GPCRs can be used to predict to which Gα protein a particular C. elegans GPCR might couple (e.g., Wragg et al., 2007). However, a more definitive assignment requires experimental evidence. Two lines of such experimental evidence are described below, and Table 1 lists the lines of evidence for Gα protein coupling available for each of 22 C. elegans receptors.
The first line of experimental evidence is listed in Table 1 as “heterologous cell” studies. Here, a GPCR is expressed in heterologous cultured cells for ligand activation studies, as described above in Section 2.1.1. In addition to identifying an activating ligand, these studies can determine what type of Gα protein is activated by the GPCR in heterologous cells. For example, activation of Ca2+ release generally indicates Gαq signaling, activation of cAMP production generally indicates Gαs activation, and inhibition of cAMP production and/or sensitivity to pertussis toxin generally indicates Gαi/o activation.
The second line of experimental evidence listed in Table 1 comes from genetic studies in C. elegans. Here, a C. elegans GPCR mutant can be used to demonstrate that a receptor acts in a particular cell type to support a particular behavior, and genetic studies can similarly show that a particular Gα protein is also required in the same cell type for the same behavior.
Table 1 shows that 20 of 22 GPCRs assigned to a ligand are also assigned coupling to a specific Gα protein, with the number of experimental lines of evidence for that Gα assignment varying from zero to two. Gα assignments from heterologous cell studies generally agree with assignments from C. elegans genetics in cases where both lines of evidence are available, bolstering confidence in assignments from the many cases with only one line of evidence. An interesting case is that of the dopamine receptor DOP-2, which was assigned to Gαi/o in heterologous cell studies, and for which C. elegans genetics suggests that three different Gαo-related Gα proteins may mediate DOP-2 signaling in worms (Suo et al., 2003; Suo et al., 2009; Correa et al., 2012; Pandey and Harbinder, 2012; Mersha et al. 2013).
At present at least 119 C. elegans genes encoding over 250 neuropeptides have been cataloged (Li and Kim, 2008; Li and Kim, 2010). Identifying the receptors for all these neuropeptides is a major challenge. Of the >1,000 putative GPCRs encoded in the C. elegans genome, most are chemosensory receptors, and a much smaller subset are likely to be neuropeptide receptors. For tables itemizing these receptors, I refer readers to several recent reviews that have cataloged the likely C. elegans neuropeptide receptors by looking for GPCRs most similar to known neuropeptide receptors (Altun, 2011; Frooninckx et al., 2012; The neuronal genome of Caenorhabditis elegans). These cataloging efforts generally predict about 150 genes encoding neuropeptide receptors, with individual genes often producing a number of differentially spliced isoforms.
Only a relatively small number of putative neuropeptide receptors have been assigned as receptors for specific neuropeptide(s)—one recent count put the number of such “deorphanized” neuropeptide receptors at 23 (Frooninckx et al., 2012). The quality of the data varies for each receptor, but a definitive assignment of a peptide to a receptor should include data showing the peptide binds the receptor with high affinity and specificity, as well as C. elegans genetic data demonstrating that the peptide functions via the receptor to regulate a specific behavior. An example of such a definitive assignment is work showing that the sensory BAG neurons release two different peptides encoded by the flp-17 gene which then act via the EGL-6 receptor on the HSN motor neuron to inhibit egg-laying behavior (Ringstad and Horvitz, 2008).
Can C. elegans neuropeptides or neuropeptide receptors be matched up with mammalian orthologs? In the case of the neuropeptides, the sequences are so short that there just is not enough information content in them to make such an analysis possible. In the case of the receptors, their information-rich sequences do allow such an analysis. Mammalian neuropeptide receptors can be broken down into several subfamilies based on sequence relationships, and many worm receptors can similarly be fitted into the same families (Altun, 2011; Frooninckx et al., 2012; The neuronal genome of Caenorhabditis elegans). Most C. elegans putative neuropeptide receptors cannot be definitively assigned as orthologs of specific mammalian receptors simply based on sequence analysis. However, based on sequence similarity as well as additional functional data, some worm receptors have been described as models for specific mammalian receptors. Thus C. elegans NPR-1 is similar to the mammalian neuropeptide Y receptor (de Bono and Bargmann, 1998), C. elegans PDFR-1 is similar to the Drosophila pigment dispersing factor receptor and mammalian vasoactive peptide and calcitonin receptors (Janssen et al., 2008), and C. elegans NPR-17 is similar to mammalian opioid receptors (Cheong et al., 2015).
An important result from analysis of G protein coupled neurotransmitter receptors in C. elegans is that each receptor tends to be expressed on a small number of specific neurons, allowing that receptor to mediate very specific effects on behavior. C. elegans hermaphrodites have just 302 neurons, which, due to symmetries and repeated structures in the anatomy, can be classified into 118 types. Each neuron can be identified by its unique position and morphology within the animal. Further, all the synaptic connections of each neuron with other neurons and muscles have been mapped, making C. elegans the only animal for which a complete neural wiring diagram is available (White et al., 1986). Because of these features, identifying all the specific neurons that express a neurotransmitter and its receptor is uniquely possible in C. elegans, and this information, once obtained, can be interpreted using the wiring diagram to obtain unique insights into neural signaling. In this section I begin a discussion of this area of investigation.
The expression patterns of a significant subset of C. elegans neural GPCRs have been studied. These experiments generally involve creating transgenic worms in which the promoter and other regulatory sequences for a GPCR gene are used to drive expression of the green fluorescent protein (GFP), and specific cells expressing GFP are identified by fluorescence microscopy. Data for the expressions patterns of 22 GPCR receptors for small-molecule neurotransmitters are summarized in Table 2, and expression patterns for 27 of the ∼150 putative neuropeptide receptors are also available (The neuronal genome of Caenorhabditis elegans). The quality and completeness of the work varies greatly for each receptor. In some cases, only relatively short promoter regions were used to construct the GFP reporter transgenes, raising questions as to whether the results obtained fully represent the expression patterns of the endogenous GPCR genes. In other cases, only a cursory analysis of the fluorescently labeled cells is presented, with little or no attempt to identify the GFP-expressing cells.
Despite these caveats, an important generalization can be made about the expression patterns of neural GPCRs: an individual neurotransmitter receptor type tends to be expressed in a limited set of specific cells. This can be illustrated with examples from receptors for which relatively high-quality expression data are available and which typify the results obtained with other receptors. A GFP reporter for the EGL-6 neuropeptide receptor was expressed in just three types of neurons (HSN, SDQ, DVA) and one type of glial cell (GLR) (Ringstad and Horvitz, 2008). A GFP reporter for the NPR-1 neuropeptide receptor was expressed in approximately 20 neuron cell types (Coates and de Bono, 2002). A reporter for the NPR-4 neuropeptide receptor was expressed in five neuron types, the intestine, and the rectal gland cell, while a reporter for the NPR-5 neuropeptide receptor was expressed 13 neuron types and in all body wall muscles (Cohen et al., 2009). Expression patterns of G protein coupled receptors for small-molecule neurotransmitters (Table 1) show similar characteristics to those seen in the above examples of neuropeptide receptors—each receptor is typically expressed in a limited number of neuron types plus sometimes additional non-neuronal cell types. For example, a reporter for the TYRA-2 tyramine receptor is expressed in about 14 neuron types (Rex et al., 2005). Reporters for the SER-2 tyramine receptor are expressed in a total of 24 neuron types, plus body wall muscles, pharyngeal muscles, and the excretory gland (Tsalik et al., 2003).
The predominant model of neural signaling has been that synapses, specialized physical connections between neurons, are the sites of neurotransmitter signaling between neurons. A presynaptic neuron releases neurotransmitter from vesicles clustered at a presynaptic terminus, flooding the narrow synaptic cleft with neurotransmitter, which binds to neurotransmitter receptors clustered on the postsynaptic membrane. The idea that synapses are central to understanding neural function inspired the landmark achievement of mapping all the synaptic connections in the C. elegans nervous system (White et al., 1986), and is behind current efforts, for example within the BRAIN initiative, to map synaptic connections within more complex nervous systems (Jorgenson et al., 2015).
While ionotropic neurotransmitter signaling may typically be restricted to synapses, studies in C. elegans provide ample evidence that neurotransmitter signaling through G protein coupled receptors may be predominantly extrasynaptic. The concept of extrasynaptic neurotransmission (also called volume transmission) originated in 1986 based on studies of mammalian brain (Agnati et al., 1986). Since then, many studies in mammalian brain demonstrated the release of neurotransmitters from extrasynaptic sites, the ability of neurotransmitters to diffuse through the extracellular space, and the localization of G protein coupled neurotransmitter receptors to extrasynaptic sites (Agnati et al., 2010). Indeed, given the submicromolar affinities of many GPCRs for their neurotransmitter ligands (see Table 1 for examples), it would make little sense for such receptors to function at synapses where neurotransmitter concentrations can rise to the millimolar level (Barberis et al., 2011) and may never be reduced to low enough concentrations to allow high-affinity receptors to become unliganded. Despite this evidence, it has been difficult to provide genetic evidence demonstrating the functional significance of extrasynaptic signaling in mammalian brain. In contrast, C. elegans is ideally suited to such work. In this system, it is possible to use rigorous genetic experiments to define the specific neurons that release a neurotransmitter to induce a particular behavioral response, and to also define the specific neurons that express the G protein coupled receptor that functionally receives the neurotransmitter signal to execute the response. Remarkably, results of such analyses show over and over again that the neurotransmitter releasing neuron and the receptor-expressing receiving neuron are not synaptically connected. Ironically, the complete synaptic wiring diagram of the C. elegans nervous system has thus been the key tool for demonstrating that synapses are often not needed to mediate neurotransmitter signaling via G protein coupled receptors. Here I summarize the evidence for extrasynaptic neurotransmitter signaling in C. elegans.
Neuropeptides and some small-molecule neurotransmitters are released from dense-core vesicles, which are distinct from the small-clear vesicles clustered at presynaptic termini. In C. elegans neurons that make synapses dense-core vesicles are excluded from the synaptic active zones where synaptic vesicles are released, although many are localized nearby (Hammarlund et al., 2008 ). Furthermore, C. elegans, like the human brain, has specialized neuroendocrine cells that synthesize and release neurotransmitters but that do not form synaptic connections with any other cells. The uv1 neuroendocrine cells release tyramine and neuropeptides to inhibit egg-laying behavior (Jose et al., 2007), and the NSM neuroendocrine cells release serotonin to regulate locomotion (Sawin et al., 2000; Gürel et al., 2012). The extrasynaptic neurotransmitter release sites of NSM have been studied in some detail (Nelson and Colón-Ramos, 2012). The documented functions of neurotransmitter release from uv1 and NSM provide one set of evidence that neurotransmitters do signal extrasynaptically. Some additional cells in C. elegans do not form any synapses, yet express neuropeptide genes, so any functions of peptides secreted from these cells must also be extrasynaptic. Cells in this category include the CAN cell, intestinal cells, muscle cells, and hypodermal cells (White et al., 1986; Li and Kim, 2008; Li and Kim, 2010).
The specific C. elegans neurons that synthesize and release individual small molecule neurotransmitters or neuropeptides have been mapped in considerable detail (see WormBook chapter GABA; Rand, 2007; Chase and Koelle, 2007; Li and Kim, 2008; Li and Kim, 2010; Jafari et al., 2011; Serrano-Saiz et al., 2013; Periera et al., 2015). Thus when the expression pattern of a G protein coupled receptor for a specific neurotransmitter is determined, we can use the synaptic wiring diagram of C. elegans to see if the cells that produce that neurotransmitter are presynaptic to the cells that express its receptor. In many cases, the answer is no.
G protein coupled receptors for small-molecule neurotransmitters are expressed on cells not postsynaptic to neurons that release the corresponding neurotransmitter. This was first noted for the dopamine receptors DOP-1 and DOP-3, which are expressed, among other places, on ventral cord motor neurons (Chase et al., 2004). Dopamine is released from exactly three types of neurons in C. elegans, and none of these neurons make synapses onto the ventral cord motor neurons. Genetic studies show that dopamine regulates locomotion via the DOP-1 and DOP-3 receptors, and the locomotion defects of dop-1 and dop-3 mutants can be rescued be re-expressing the receptors specifically in ventral cord motor neurons (Chase et al., 2004). These experiments rigorously demonstrate that dopamine receptors are not only found on cells distant from dopamine release sites, but that they also function to mediate dopamine signaling in these distant cells.
Since those initial studies of dopamine signaling, similar experiments have demonstrated extrasynaptic signaling by other small-molecule neurotransmitters. One striking case is that of the neurotransmitter tyramine, which is released from only the neuroendocrine uv1 cells (which make no synapses) and from the RIM interneuron, which makes synapses only onto four other types of neurons and onto neck muscles (White et al., 1986). When C. elegans is touched on the head, tyramine released from RIM mediates a complex escape response. One aspect of this response is mediated by tyramine signaling at synapses from RIM onto neck muscles using an ionotropic receptor (Pirri et al., 2009). However, another aspect of the escape response is mediated by tyramine signaling from RIM through a G protein coupled receptor. This GPCR, SER-2, is expressed on GABAergic motor neurons, which are not post-synaptic to RIM, and defects in the escape response seen in ser-2 mutants are rescued by cell-specific re-expression of ser-2 in GABAergic motor neurons, demonstrating that SER-2 acts in these neurons to mediate extrasynaptic signaling by tyramine that originates from RIM (Donnelly et al., 2013).
There have now been similar genetic demonstrations that neuropeptides signal onto cells not postsynaptic to the neurons that release them. For example, FLP-17 neuropeptides are released from BAG sensory neurons and signal via the EGL-6 neuropeptide receptor on the HSN motor neurons, which are not postsynaptic to BAG, to inhibit egg laying (Ringstad and Horvitz, 2008). The C. elegans defecation motor program is coordinated by NLP-40 peptides released from intestinal cells, which being non-neuronal make no synapses, and these NLP-40 peptides signal onto GABAergic motor neurons via the AEX-2 peptide receptor (Wang et al., 2013). Beyond these rigorous genetic demonstrations that specific peptides signal extrasynaptically via particular neuropeptide receptors, there are additional data showing that some neuropeptide receptors are expressed on cells that do not receive any synapses, so that any signaling they mediate on these cells must be extrasynaptic. These receptor-expressing cells include intestinal, hypodermal, and glial cells (The neuronal genome of Caenorhabditis elegans).
Null mutants or RNAi knockdowns for many C. elegans G protein coupled neurotransmitter receptors have been analyzed, and the main finding is that phenotypic defects are very hard to detect in them. Most of the time no defects are observed, and the few mutant phenotypes that have been described for neural GPCRs are typically not gross behavioral defects obvious upon inspection of individual animals, but rather narrow behavioral defects detectable only when populations of worms are put through specific phenotypic assays.
An initial effort at identifying neural GPCR phenotypes used RNAi to knock down expression of 60 G protein coupled neurotransmitter receptors (Keating et al., 2003) and simply looked for uncoordinated movement or defects in reproduction. Knockdown of seven of the receptors had detectable effects on the frequency and/or amplitude of body bends, while knockdown of five receptors had some effect on the production of progeny. Later large-scale RNAi screens for genes that affect acetylcholine release/signaling identified hits in five GPCR genes, of which three were validated using genetic knockout mutations (Sieburth et al., 2005; Vashlishan et al., 2008). It is difficult to interpret some of these RNAi results: not all the defects seen with RNAi were reproduced with genetic mutations, the RNAi results did not always reproduce in different labs, and the phenotypes that were observed have not been characterized in much detail. Negative results from RNAi for GPCRs are also difficult to interpret since gene expression in C. elegans neurons is not always efficiently knocked down by RNAi, even in mutant backgrounds that enhance neuronal RNAi (Poole et al., 2011).
Mutations in genes encoding GPCRs have almost never been generated in forward genetic screens, despite the large number of GPCR genes in the worm genome. Since Brenner’s first screen for uncoordinated mutants (Brenner, 1974), C. elegans mutants for thousands of genes have been generated in genetic screens, including a large number with defects in neural structure and/or function. Included in this trove are many mutants for heterotrimeric G proteins and other signaling proteins that act downstream of neural GPCRs (Perez-Mansilla and Nurrish, 2009), making the absence of neural GPCR mutants even more conspicuous.
Considering the few neural GPCR mutants that have arisen in forward genetic screens is instructive. The EGL-6 neuropeptide receptor was identified by a rare gain-of-function mutation that inhibits egg laying (by increasing EGL-6 signaling), yet knockout mutations of EGL-6 do not produce detectable egg-laying defects (Ringstad and Horvitz, 2008). Loss-of-function mutations in G protein coupled serotonin and dopamine receptors have arisen in forward genetic screens, but in these cases the phenotype screened for was resistance to the paralysis induced by applying these neurotransmitters to worms at high concentrations (Chase et al., 2004; Gürel et al., 2012). These mutants do not have obvious behavioral defects when animals are not bathed in a neurotransmitter, although defects have been detected when populations of mutant animals are put through very specific behavioral assays known to depend on serotonin or dopamine. There is perhaps only a single example of a neural GPCR for which loss-of-function mutants have been recovered from a forward genetic screen and for which the mutant phenotype is easily observable in single animals: mutants lacking AEX-2, the receptor for NLP-40 neuropeptides, are defective in the expulsion step of defecation (Mahoney et al., 2008 ; Wang et al., 2013).
C. elegans mutants for many G protein coupled neurotransmitter receptors are available as a result of large-scale efforts to produce gene knockouts for all C. elegans genes (C. elegans deletion mutant consortium, 2012). However, phenotypic defects have been described for only a small subset of these GPCR mutants. The cases in which phenotypic defects have been detected have generally come when either the neurotransmitter that activates the GPCR was known, so that specialized assays for behaviors dependent on that neurotransmitter could be assayed for defects, or in cases in which the expression pattern of the GPCR was known, so that specialized assays for behaviors dependent on neuron(s) that express the receptor could be tested for defects. An example that illustrates both strategies comes from the work of Harris et al. (2009), which analyzed how C. elegans backs away from the odor of dilute octanol, a response known to depend on both the neurotransmitter serotonin and on a neural circuit containing specific sensory neurons and interneurons. This information was used to focus in on two G protein coupled serotonin receptors, one expressed in the interneurons, another in the sensory neurons, and further experiments showed that knockouts and knock downs of these receptors resulted in specific behavioral defects in response to octanol. The two serotonin receptors studied by Harris et al. (2009) are part of the set of 22 receptors for small molecule neurotransmitters listed in Table 2, which are the most intensively studied GPCRs in C. elegans. As shown in Table 2, at this point mutant defects have been described for 20 of these receptors. The situation is less encouraging in the case of neuropeptide receptors, with mutant phenotypic defects described for only a few out of this large set of ∼150 receptors.
Considering the totality of available genetic studies of C. elegans G protein coupled neurotransmitter receptors, it appears that the knockout phenotypes for these proteins are generally so narrow that they will not be easily detected. Because individual receptors are expressed in only a few types of neurons, it is reasonable to expect that just the very specific behaviors affected by those neurons will be affected, and that specialized behavioral assays will be required to detect these defects. Another possible reason that GPCR mutant phenotypic defects are rarely detected could be that the large family of GPCRs contains many functionally redundant receptors, such that knocking out one receptor will not give obvious defects unless its redundant partners are also knocked out simultaneously. Several examples support the idea that neurotransmitter receptors can function redundantly. Two serotonin receptors appear to be co-expressed on the vulval muscles and both appear to promote activity of these muscles (Hapiak et al., 2009). Serotonin released from the NSM neurosecretory cells inhibits locomotion, and two different serotonin receptors expressed on largely non-overlapping sets of neurons redundantly mediate this effect (Gürel et al., 2012). Neuropeptides encoded by FLP-18 modulate locomotion, and three different receptors, NPR-1, NPR-4, and NPR-5, expressed in different sets of cells, redundantly mediate these effects of FLP-18 (Stawicki et al., 2013). FLP-18 signaling is quite complex, because other behavioral effects of FLP-18 peptides are mediated non-redundantly by NPR-1, and for these effects FLP-18 and FLP-21 neuropeptides both seem to activate NPR-1 (Choi et al., 2013). If such complex functional relationships between neurotransmitters and receptors are widespread, this will make assigning functions to receptors using knockout mutations very challenging.
A tool that may prove generally useful in genetically characterizing redundant or subtle GPCR functions is transgenic overexpression of GPCRs. One example comes from studies of the EGL-6 neuropeptide receptor. Loss-of-function mutants for EGL-6 do not result in obvious defects under standard lab growth conditions. However, using a transgene carrying many copies of egl-6 genomic DNA to presumably overexpress the receptor does result in an obvious egg-laying defective phenotype, apparently due to increased activation of the overexpressed EGL-6 receptor by the FLP-10 and FLP-17 neuropeptides (Ringstad and Horvitz, 2008). Another example comes from studies of the NPR-17 neuropeptide receptor. NPR-17 mediates an effect of NLP-3 peptides to modulate how C. elegans backs away from aversive stimuli, and a transgene carrying many copies of the npr-17 gene causes a gain-of-function phenotype opposite that of an nlp-3 or npr-17 knockout, and that depends on the presence of a wild-type NLP-3, suggesting that the overexpressed NPR-17 receptor causes increased NPR-17 signaling (Harris et al., 2010). If transgenically overexpressing a GPCR is generally able to induce gain-of-function phenotypes, this could be a valuable approach for identifying subtle or redundant functions of GPCRs.
A major area of current research on mammalian GPCRs concerns the homo- and hetero-oligomerization of these receptors (González-Maeso, 2011). It appears that in many cases, two different types of GPCRs can exist and function in vivo as a heterodimeric complex, and that a heteromeric receptor can have very different signaling properties than homomers of its subunits, including the ability to bind to different ligands. While GPCR heteromerization has so far been studied mainly using biochemical and biophysical methods, genetic analysis has much to contribute to understanding the physiological significance of this phenomenon. Heteromerization also potentially vastly complicates the challenge of understanding GPCR function: if C. elegans has ∼150 G protein coupled neurotransmitter receptor genes, how many types of heteromeric GPCRs might actually be the functional units in vivo?
There have so far been only a few efforts to analyze GPCR heteromer function in C. elegans. One example is that of the G protein coupled receptor for the neurotransmitter GABA. In mammals this GABAB receptor is an obligate heteromer between the GABAB1 and GABAB2 subunits (Kaupmann et al., 1998). C. elegans has orthologs of each subunit, known as GBB-1 and GBB-2, respectively. Knockouts of either GBB-1 or GBB-2 can block all G protein coupled responses to GABA in worms (Schultheis et al., 2011), and GBB-1 and GBB-2 act together to alter responses to the drug aldicarb (Dittman and Kaplan 2008; Vashlishan et al., 2008), consistent with the idea that the GABAB receptor is an obligatory heterodimer in worms just as it is in mammals. A second example comes from studies of signaling onto sensory neurons, not by a neurotransmitter, but by the mixture of small molecules known as ascarosides that collectively make up the worm dauer pheromone. The functional receptor for the specific ascaroside isoform ascr#2 appears to be a heteromer of the GPCRs DAF-37 and DAF-38 (Park et al., 2012). ascr#2 binds directly to DAF-37, and DAF-37 is essential for response to ascr#2 but is not involved in the response to other ascaroside isoforms. Genetic studies show that DAF-38 assists but is not essential for response to ascr#2, and similarly assists response to other ascaroside isoforms that do not signal through DAF-37, suggesting DAF-38 may heteromize with and assist signaling by a several different GPCRs that bind directly to different ascaroside isoforms. These studies demonstrate the potential of C. elegans genetic studies to sort out the intricacies of how GPCR subunits function together as heteromeric receptors in vivo.
One of the principal issues in studying neural GPCRs is identifying the specific neurotransmitters that activate them. At this point, 22 of 28 GPCRs encoded in the worm genome that seem likely to be receptors for small-molecule neurotransmitters have been matched with their activating ligands (Table 1), but about 85% of the ∼150 putative neuropeptide receptors remain “orphans”, that is receptors with unknown activators (Frooninckx et al., 2012).
The general strategy to deorphanize neuropeptide receptors is to express them in heterologous cells, to apply synthetic versions of each neuropeptide encoded in the genome, and to test which specific peptides can activate receptor signaling. There are a number of cell types and signaling assays that have been used successfully for this purpose (Mertens et al., 2004). This strategy has been applied to a number of C. elegans neuropeptide receptors, for example, EGL-6 (Ringstad and Horvitz, 2008) and NPR-1 (Rogers et al., 2003 Kubiak et al., 2003). The reverse strategy has also been used, screening through a set of C. elegans neuropeptide receptors to identify those that can be activated by a particular neuropeptide of interest (Cohen et al., 2009). The results of receptor deorphanization experiments are complex: often a single receptor can be activated by multiple different peptides, the same peptides can activate multiple different receptors, the results can vary depending on the cell type and assay system used, and the EC50 values measuring the potencies with which peptides activate receptors vary from the nanomolar to micromolar ranges (Peymen et al., 2014).
C. elegans provides the opportunity to analyze mutants for the receptors and peptides putatively matched by such deorphanizing experiments to determine if they actually function together in vivo. Genetic approaches can also be used to match peptides and their receptors, for example, screening RNAi knockdowns of many neuropeptide genes to find those that phenocopy a particular orphan receptor mutant (Cohen et al., 2009). Such genetic studies have generated results that are similar in complexity to those from using signaling assays in cultured cells for receptor deorphanization. Thus it appears that in vivo individual receptors can be activated by multiple peptides and individual peptides can act through multiple receptors (Choi et al., 2013; Stawicki et al., 2013).
The gold standard in matching neuropeptides with receptors is to combine studies of receptor activation by purified peptides in cultured cells with genetic studies in C. elegans. For the most part, it has been possible to achieve consistent results between the two approaches, generating strong confidence in the results, although there have been occasional puzzling exceptions (Cheong et al., 2015). Overall, the data matching neuropeptides to their cognate receptors remains sparse and we have a long way to go to fully match up these signaling molecules.
In order for signaling by an activated GPCR to terminate, the receptor must eventually be inactivated. One mechanism for terminating receptor activity is to clear neurotransmitter from the extracellular space so that it no longer remains bound to the receptor. There are enzymes that degrade neurotransmitters and transporters that take them back up into cells that function for this purpose (Zimmerman and Soreq, 2006; Kanner and Zomot, 2008). Another mechanism for terminating signaling involves G protein coupled receptor kinases (GRKs) that specifically recognize and phosphorylate active GPCRs, and arrestin proteins that bind phosphorylated receptors. Phosphorylation and arrestin binding can block the ability of receptors to activate G proteins and also cause cells to internalize receptors from the cell surface, down-regulating signaling (Kohout and Lefkowitz, 2003). These mechanisms, referred to as receptor desensitization, have been extensively studied in mammalian cells. Studies in C. elegans of GRK and arrestin homologs have identified specific functions of these proteins in sensory neurons (Fukuto et al., 2004; Palmitessa et al., 2005, Pereira and van der Kooy, 2012; Singh and Aballay, 2012), but have so far failed to show any striking effects of GRK or arrestin on neurotransmitter signaling in the rest of the nervous system.
It is interesting to extrapolate from the existing data on C. elegans G protein coupled neurotransmitter receptors to consider what the overall landscape of neural signaling through GPCRs may look like. If we imagine that the ∼175 neural GPCRs in C. elegans are each expressed on average in 10 of the 118 neural cell types, then a typical neuron would express about 15 GPCRs, about two of which might be small-molecule neurotransmitter receptors, with the remainder being neuropeptide receptors. This typical neuron would then face the task of simultaneously sensing the levels of ∼15 neurotransmitters in its extracellular space and executing appropriate responses to the dynamic mix of these signals it receives over time. All the GPCRs in this typical neuron may signal through about three types of Gα proteins, which do not act separately but rather collaborate to modulate neurotransmitter release from the neuron (see Section 3). Thinking about neurotransmitter signaling through GPCRs this way, it does not make sense to consider the action of a single neurotransmitter at a time, which is the way that we currently investigate neural signaling. Rather, it may be more appropriate to investigate how a single neuron computes appropriate responses to the entire mix of neurotransmitters in its environment. A prerequisite to such an investigation would be to know all the GPCRs that are present on that neuron, and we currently do not have that information for even a single C. elegans neuron.
Perhaps the most striking result from analysis of C. elegans neural GPCRs is that neurotransmitters signal extrasynaptically through these receptors. Thus neurons that have no physical connections can signal each other and work together to control specific behaviors. The predominant model for understanding nervous system function has been that the functional ensembles of neurons that control thoughts and behaviors are circuits defined by the synaptic connections between the neurons in the ensemble. We have had the synaptic wiring diagram for the C. elegans nervous system for almost 30 years (White et al., 1986) and it has proven insufficient to allow us to understand the neural control of behaviors in this organism. Now that we understand the widespread nature of extrasynaptic neurotransmitter signaling, we need to expand our understanding of a neural circuit to be a functional ensemble of neurons that signal each other but that may lack direct anatomical connections. The patterns of signaling via GPCRs are not determined by the synaptic wiring of the nervous system, but rather by the specific expression patterns of neurotransmitters and their GPCR receptors. Thus to help understand neural circuit function, we need to supplement the existing synaptic wiring diagram with an additional diagram in which the specific cells that express each neurotransmitter and its cognate receptor(s) are defined. This goal, while ambitious, is potentially achievable in the C. elegans system, perhaps aided by new cell-specific RNAseq technologies (Spencer et al., 2014).
In this section, I focus on genetic studies in C. elegans of signaling by the neural G proteins Gαo, Gαq, and Gαs that mediate signaling by G protein coupled neurotransmitter receptors. The G proteins themselves and their downstream signaling pathways are strongly conserved comparing mammalian brain and C. elegans. Genetic studies in C. elegans show that all three types of Gα proteins signal to regulate neurotransmitter release. C. elegans genetic screens have been used to discover new signaling molecules that regulate neural G protein signaling, including Regulators of G protein Signaling (RGS proteins) that help terminate signaling, the RIC-8 protein that is required for signaling, and a new Gαq effector (Trio’s RhoGEF domain). The in vivo studies of neural signaling in C. elegans suggest a model in which the heterotrimeric G proteins present in a neuron act to integrate signaling through the multiple GPCRs present on the neuron to produce its appropriate output level.
I will focus here on the G protein subunits that mediate neurotransmitter signaling, and refer readers to an earlier review for a table listing all Gα, Gβ, and Gγ subunits in C. elegans, and for discussion of the large family of specialized C. elegans Gα proteins found in sensory neurons that mediate chemosensation (see WormBook chapter Heterotrimeric G proteins in C. elegans ).
In C. elegans, as in mammals, there are multiple G protein α, β, and γ subunits that can potentially combine to form a larger number of heterotrimeric combinations. In mammals there are few functional differences between the various β and γ subunits (Khan et al., 2013). In C. elegans only one β subunit homolog is actually used in G protein heterotrimers (Zwaal et al., 1996; Chase et al., 2001; Robatzek et al., 2001; van der Linden et al., 2001), and only one γ subunit is expressed outside the chemosensory neurons (Jansen et al. 2002). Thus in C. elegans, as in mammals, functional diversity among G protein heterotrimers rests primarily on which α subunit is used. Mammals have multiple members of each of four families of α subunits, while C. elegans has just one member for each of these four α families (Jansen et al., 1999). The four conserved C. elegans α subunits, with their corresponding mammalian orthologs and percent sequence identities to them, are: GOA-1 (Gαo, >80%), EGL-30 (Gαq, >80%), GSA-1 (Gαs, 66%), and GPA-12 (Gα12, 52%). These worm Gα proteins are each more similar to their mammalian orthologs than they are to each other. Below I will use the mammalian names to refer to the worm Gα proteins, for example using Gαo to refer to its C. elegans ortholog GOA-1. The worm Gαo, Gαq, and Gαs proteins are each widely expressed in most or all neurons, plus some muscle and other cells (Mendel et al., 1995; Ségalat et al., 1995; Park et al., 1997; Korswagen et al., 1997; Lackner et al., 1999; Bastiani et al., 2003). Gα12, in contrast, is expressed in only a small subset of neurons plus some muscle and hypodermal cells (van der Linden et al., 2003; Yau et al., 2003).
I note as an aside that while GOA-1 appears to be the Gαo ortholog in C. elegans, there are also two more distantly related Gαo homologs that have some functional redundancy with GOA-1. GPA-16 is co-expressed with GOA-1 in some neurons (Jansen et al., 1999) and in early embryonic cells, where these two Gα proteins function redundantly to control mitotic spindle positioning during asymmetric cell divisions (reviewed in Polarity establishment, asymmetric division and segregation of fate determinants in early C. elegans embryos ). GPA-7 is another Gαo-related protein that shows expression in many neurons (Jansen et al., 1999). The functions of GPA-7 have not been carefully investigated, but in one study GPA-7 and GOA-1 were shown to function redundantly to mediate dopamine signaling in C. elegans male copulatory neurons and muscles (Correa et al., 2012; ).
Studies of mammalian GPCRs demonstrate that any one receptor may activate Gαo, Gαq, Gαs, or Gα12, but will generally not couple to more than one of these Gα types (Moreira, 2014). Currently it is not possible to predict from the sequence of a GPCR which Gα protein it will activate, so this must be determined experimentally (see Section 2.1.2).
C. elegans Gα12, like mammalian Gα12, can activate its effector protein RGS-RhoGEF protein (RHGF-1 in C. elegans) to in turn activate the small GTPase Rho, and genetic work in C. elegans shows this can regulate a pathway involving diacylglycerol and protein kinase C to increase neurotransmitter release (van der Linden et al., 2003; Yau et al., 2003; Hiley et al., 2006). However, these results all arise from studies of worms expressing constitutively active mutants of Gα12, and no defects have yet been observed in worms carrying loss of function mutations in Gα12 or its effector RHGF-1. Thus the normal physiological functions of the Gα12 pathway in C. elegans remain to be elucidated. I will not further consider Gα12 signaling here, but see Perez-Mansilla and Nurrish (2009)) for a detailed review of C. elegans Gα12 signaling.
Before delving into the details of the signaling pathways for the three major neural G proteins, I begin with an overview of what these pathways do and a description of the genetic approaches in C. elegans that have been used to study them.
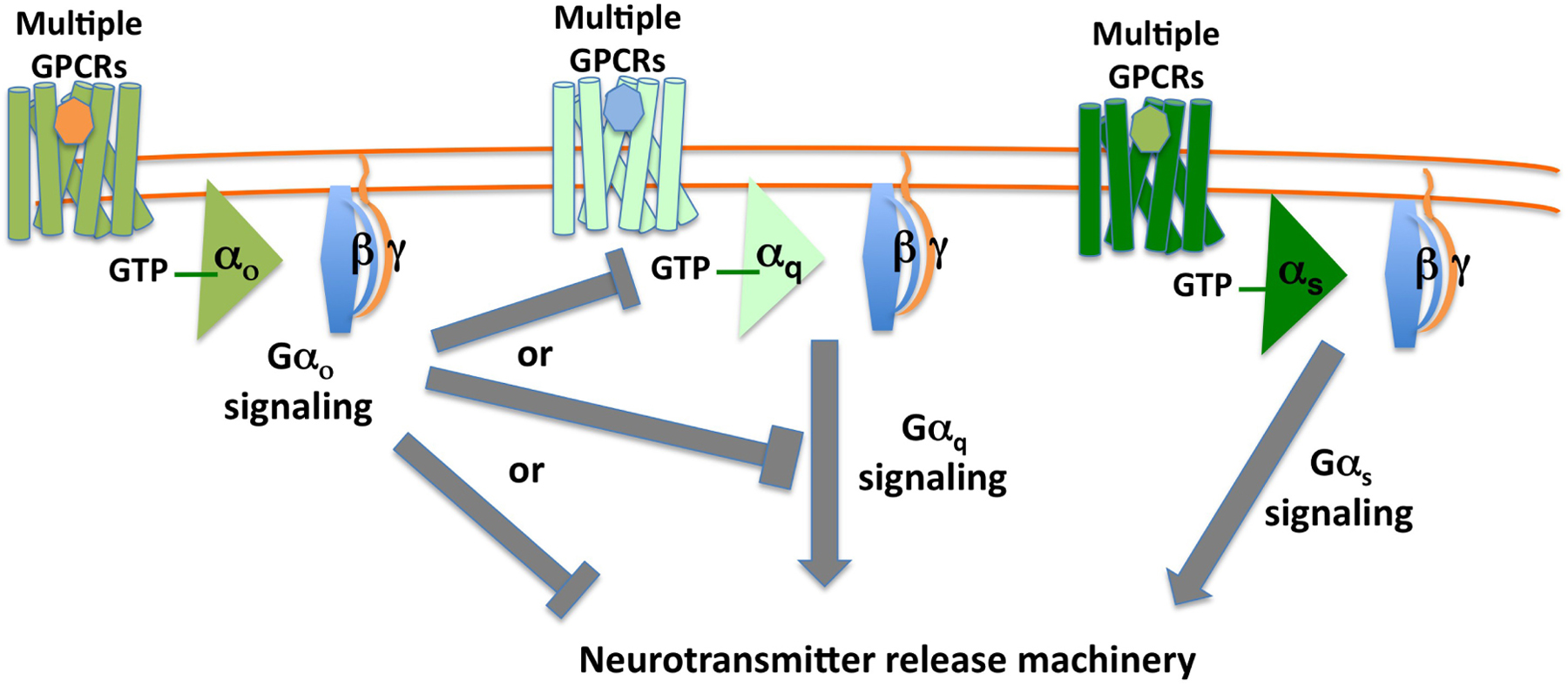
The simplified take-home message from genetic analysis of Gαo, Gαq, and Gαs signaling in C. elegans neurons is that Gαo signaling inhibits neurotransmitter release, while Gαq and Gαs signaling activate neurotransmitter release or promote synaptic activity (Figure 2). Gαo and Gαq signaling appear to affect the localization of specific molecules at presynaptic release sites that regulate the neurotransmitter release machinery itself (Lackner et al., 1999; Nurrish et al., 1999; Chan et al., 2012), while it remains less clear how Gαs signaling promotes neurotransmitter release (Reynolds et al., 2005). A single neuron expresses many GPCRs (see Section 2), so at any one time all three pathways may be active simultaneously, and indeed there may be receptors for different neurotransmitters activating a single type of Gα protein simultaneously. Thus Gαo, Gαq, and Gαs sum up signaling by the several GPCRs active on a neuron, and their three downstream signaling pathways function together to compute an appropriate efficiency for the neurotransmitter release machinery in the neuron.
|
Figure 2. Schematic diagram illustrating how Gαo, Gαq, and Gαs signaling act in a single neuron to together regulate neurotransmitter release. A schematic summary of the effects of signaling by the three Gα proteins on neurotransmitter release, as predicted from genetic studies in C. elegans. In such diagrams, an arrow indicates promotion or activation of a target, while a bar denotes an inhibitory effect. The bars extending from Gαo indicate that genetic experiments show Gαo signaling inhibits Gαq signaling, but do not determine whether this inhibition occurs upstream of Gαq, at the level of Gαq, or at some level downstream of Gαo.
Figure 2 is a gross simplification. The neurotransmitter release machinery is complex, with distinct pools of small-clear vesicles present at presynaptic termini, as well as dense-core vesicles at non-synaptic sites, and release can occur tonically or after being evoked by depolarization. Gα signaling may differentially affect the various types of vesicle release (Hu et al., 2015). Further, Gαo, Gαq, and Gαs signaling are known from electrophysiological studies to affect activity of specific ion channels, and from studies in other experimental systems to affect gene expression and synaptic structure. These G proteins are expressed not only in neurons but also in other cell types (e.g., muscles). So, clearly signaling by Gαo, Gαq, and Gαs must have effects other than on the neurotransmitter release machinery. Despite this, studies in C. elegans have focused on the effects of G protein signaling on synaptic neurotransmitter release because genetic experiments demonstrate that they actually affect behavior.
There are multiple lines of evidence that Gαo, Gαq, and Gαs signaling affect neurotransmitter release in C. elegans, with the most extensive evidence coming from studies of acetylcholine release by ventral chord motor neurons that control locomotion behavior. The four major lines of evidence are: 1) mutations in the G proteins and/or their signaling pathways alter locomotion behavior; 2) mutations in the G proteins and/or their signaling pathways alter response to aldicarb, an inhibitor of the acetylcholinesterase enzyme that clears released acetylcholine from synapses; 3) mutations in the G proteins and/or their signaling pathways alter the localization of GFP-tagged presynaptic proteins at cholinergic synapses; and 4) mutations in the G proteins and/or their signaling pathways cause changes in acetylcholine release that can be measured with electrophysiological methods. The specific studies detailing these lines of evidence for Gαo, Gαq, and Gαs are described and cited below in the remainder of Section 3. As these details will show, all four lines of experimental evidence have established the effects of Gαo and Gαq signaling on acetylcholine release in C. elegans, while the effects of Gαs signaling on acetylcholine release from C. elegans ventral cord motor neurons rest on just the first two lines of evidence. However additional lines of evidence for the effects of Gαs signaling on neurotransmitter release come from studies of other cell types and species. For example studies in C. elegans ALA neurons show that the Gαs signaling pathway affects dense-core vesicle release (Zhou et al., 2007), and electrophysiological studies in mammalian and Drosophila neurons have also established effects of Gαs signaling on release of small clear neurotransmitter vesicles (Trudeau et al., 1996; Chen and Regehr, 1997; Kuromi and Kidokoro, 2000).
Studies of C. elegans neural G protein signaling have been carried out in parallel through both forward and reverse genetics. In the reverse genetic approach, C. elegans homologs of mammalian signaling proteins are knocked out by gene-targeting technologies. Thus, for example, the entire sets of G protein subunit genes and RGS genes have been knocked out and analyzed (Jansen et al., 1999; Hess et al., 2004). The special power of the C. elegans system, however, is its capacity for large-scale forward genetic screens in which the genome is randomly mutagenized and vast numbers of animals are screened for specific phenotypes associated with neural G protein signaling defects. The initial screens for mutants with general neural G protein signaling defects took advantage of the fact that heterotrimeric G proteins regulate neurotransmitter release from the egg-laying motor neurons and from the cholinergic motor neurons that control locomotion. Thus these screens looked for mutants that fail to lay eggs (Trent et al., 1983; Desai and Horvitz, 1989), that lay eggs too frequently (Bany et al., 2003), that alter acetylcholine release (Miller et al., 1996; Miller et al., 1999; Miller et al., 2000; Sieburth et al., 2005; Vashlishan et al., 2008), that carry suppressors of previously-isolated G protein signaling mutations (Miller et al., 1999; Schade et al., 2005; Charlie et al., 2006b; Williams et al., 2007), or that have hyperactive locomotion (Schade et al., 2005). Additional screens were for mutants that fail to respond to specific neurotransmitters that signal through GPCRs, including dopamine (Chase et al., 2004: ) and serotonin (Gürel et al., 2012).
All genetic studies of neural G protein signaling in C. elegans involve assaying mutant phenotypes that arise from G protein signaling defects. Here I describe some of the mutant phenotypes used for this work.
Gαo null and partial loss-of-function mutants have a “hyperactive” phenotype (Mendel et al., 1995; Ségalat et al., 1995), which, as described below, appears to arise from increased neurotransmitter release from neurons throughout the animal. This phenotype can easily be recognized by observing animals growing on a standard laboratory petri dish (Movie 1) and includes body bends that are deeper and more frequent than in the wild type. This defect arises at least in part from increased release of acetylcholine from the ventral cord motor neurons that control locomotion (Vashlishan et al., 2008). The hyperactive phenotype also includes an increased frequency of egg-laying behavior, such that animals lay almost all their eggs as soon as they are produced, so that whereas a wild-type animal might carry ∼12 unlaid eggs on average, a hyperactive mutant might carry only one or two (Movie 1). This defect arises at least in part from increased neurotransmitter release from the HSN motor neurons that stimulate egg laying (Tanis et al., 2008). Specific serotonin and dopamine receptors signal through Gαo to inhibit locomotion, such that treating worms with sufficiently high concentrations of dopamine or serotonin paralyzes wild-type worms, but hyperactive Gαo mutants are resistant to paralysis by serotonin or dopamine (Ségalat et al., 1995; Chase et al., 2004; Gürel et al., 2012). As might be expected, since Gαo is expressed in many neurons and non-neuronal cells, Gαo mutants have a number of additional phenotypic defects, including defects in mitotic spindle movements in early embryonic cells (Miller and Rand, 2000; Gotta and Ahringer, 2001) and defects in meiotic maturation of germ cells (Govindan et al., 2006).
|
Movie 1. The opposite effects of Gαo and Gαq loss-of-function mutations on behavior. A-C, still images of adult mid-body regions of wild type, goa-1(n1134) Gαo partial loss-of-function mutant, and egl-30(n686) Gαq partial loss-of-function mutant animals, respectively. Unlaid eggs are visible as oval objects inside the body. While wild-type animals average ∼12-16 unlaid eggs, Gαo mutants lay almost all their eggs and thus accumulate only 2-3 unlaid eggs, while Gαq mutants fail to lay eggs and accumulate <40 unlaid eggs. D-F, videos of locomotion by larvae of the same genotypes. The locomotion defects seen are most obvious in larvae but adults show qualitatively similar defects. Gαo mutants show hyperactive locomotion, with more frequent and deeper body bends, and more frequent reversals. Gαq mutants show sluggish locomotion, with infrequent, shallow body bends. Magnification is the same in all images, so the scale bar in B applies to all panels.
Gαq null mutants undergo developmental arrest as young larvae (Reynolds et al., 2005), but partial loss-of-function mutants develop to adulthood and show a “sluggish” phenotype that appears to arise from decreased neurotransmitter release (Lackner et al., 1999; Miller et al., 1999) from neurons throughout the animal (Bastiani et al., 2003). Sluggish animals have less frequent and shallower body bends than in the wild type, and strong loss-of-function or null Gαq mutants are virtually paralyzed (Movie 1) (Reynolds et al., 2005; Williams et al., 2007). This defect results at least in part from decreased acetylcholine release from ventral cord motor neurons (Lackner et al., 1999; Hu et al., 2015). Sluggish animals rarely engage in egg-laying behavior, such that adult animals can become bloated with up to ∼50 unlaid eggs (Movie 1), and this defect arises at least in part from decreased neurotransmitter release from the HSN motor neurons (Tanis et al., 2008).
The Gαo and Gαq loss-of-function phenotypes are in many respects precisely the opposite of each other. This interpretation is reinforced by the fact that mutations and transgenes that increase Gαo signaling cause a sluggish phenotype essentially indistinguishable from that seen in Gαq loss-of-function mutants (Mendel et al., 1995; Ségalat et al., 1995; Koelle and Horvitz, 1996), while transgenes and mutations that increase Gαq signaling cause a hyperactive phenotype virtually indistinguishable from that seen in Gαo loss-of-function mutants (Bastiani et al., 2003; Hajdu-Cronin et al., 1999; Schade et al., 2005; Reynolds et al., 2005; Charlie et al., 2006a; Williams et al., 2007; Matsuki et al., 2006).
Gαs null mutations are lethal (Korswagen et al., 1997), but mutations that decrease Gαs signaling cause a sluggish, near-paralyzed locomotion phenotype similar to that of Gαq loss-of-function mutants (Moorman and Plasterk, 2002; Reynolds et al., 2005). Mutations and transgenes that increase Gαs signaling induce a smoothly sinusoidal hyperactive phenotype, distinctly different from the deep body bends seen in Gαo loss-of-function or Gαq gain-of-function mutants (Reynolds et al., 2005; Schade et al., 2005; Charlie et al., 2006a).
While Gαo, Gαq, and Gαs signaling affect many C. elegans behaviors, locomotion and egg-laying behaviors are two readouts of neural G protein signaling that have been frequently used to carry out genetic studies of the mechanism of neural G protein signaling due to the unique advantages of each.
Egg-laying behavior depends on release of serotonin and other neurotransmitters from the HSN motor neuron. Gαo and Gαq have opposing effects on HSN neurotransmitter release (Tanis et al., 2008), and mutations in their signaling pathways result in easily scored and quantitated defects in egg laying (Chase and Koelle, 2004). Thus it has been possible to carry out genetic screens for mutants that are defective or hyperactive for egg-laying behavior to isolate mutants with defects in Gαo and Gαq signaling (Trent et al., 1983; Desai and Horvitz, 1989; Bany et al., 2003). Such mutants were used to originally discover and subsequently characterize the Regulators of G protein Signaling (RGS proteins) that inhibit most neural G protein signaling in C. elegans and in mammals (Koelle and Horvitz, 1996; Hajdu-Cronin et al., 1999). The neural circuit that controls egg laying is particularly simple and well-characterized, and tools to express transgenes in any cell of the circuit, to monitor activity of the circuit in freely-behaving animals with the fluorescent Ca2+ indicator GCaMP, and to optogenetically manipulate the circuit have all been developed (Schafer, 2006; Emtage et al., 2012; Collins and Koelle, 2013).
C. elegans locomotion behavior involves release of acetylcholine from ventral cord motor neurons onto body wall muscles, and Gαo, Gαq, and Gαs all affect acetylcholine release at this neuromuscular junction. The drug aldicarb paralyzes worms by preventing acetylcholine released at this neuromuscular junction from being degraded, and a powerful genetic screen for mutants resistant to aldicarb paralysis has been used to isolate mutants with decreased acetylcholine release that lie in the Gαo and Gαq signaling pathways (Miller et al., 1996; Miller et al., 1999; Miller et al., 2000). Screens for suppressors of aldicarb-resistant mutants and screens for mutants with hyperactive locomotion were used to isolate mutants with increased signaling in the Gαs and Gαq pathways (Miller et al., 1999; Schade et al., 2005; Charlie et al., 2006b). Mutations that affect acetylcholine release were used to discover the RIC-8 protein that promotes signaling by heterotrimeric G proteins in C. elegans and in mammals (Miller et al., 2000). The cholinergic neuromuscular junctions in the ventral cord have been used for important experiments visualizing the effects of G protein signaling mutations on the localization of synaptic vesicle release proteins (Lackner et al., 1999; Nurrish et al., 1999; Chan et al., 2012). These neuromuscular junctions are also the most accessible synapses for electrophysiological studies in C. elegans, and have allowed studies of the fine details of G protein signaling mutations on neurotransmitter release (Hu et al., 2015).
While mutations in the Gαo, Gαq and Gαs signaling pathways have powerful effects on egg-laying behavior by affecting neurotransmitter release from the HSN neurons and on locomotion by affecting acetylcholine release from ventral cord motor neurons, it has been hard to identify the GPCRs that activate the G proteins in these neurons. This difficulty is in line with the understanding, described in Section 2, that an individual neuron appears to express many different GPCRs so that mutations in a single GPCR are expected to have very weak effects compared to mutations in a G protein. In the HSN neuron, the EGL-6 neuropeptide receptor signals through Gαo to inhibit egg laying, but this effect is only obvious in gain-of-function EGL-6 mutants, whereas EGL-6 null mutants do not have detectable defects. In the ventral cord motor neurons, the neuropeptide receptor CKR-2 and the G protein coupled acetylcholine receptor GAR-3 both appear to signal through Gαq to increase acetylcholine release (Hu et al., 2011; Chan J.P. et al., 2013), while a different G protein coupled acetylcholine receptor GAR-2, the heterodimeric G protein coupled GABAB receptor GBB-1/GBB-2, and the dopamine receptor DOP-3 all appear to signal through Gαo to decrease acetylcholine release (Chase et al., 2004; Dittman and Kaplan, 2008). Mutations in these ventral cord motor neuron GPCRs have detectable effects on locomotion (Chase et al., 2004; Dittman and Kaplan, 2008; Hu et al., 2011; Chan J.P. et al., 2013), but knocking out a single one of these GPCRs, as expected, has much weaker effects than does knocking out its downstream G protein (Mendel et al., 1995; Ségalat et al., 1995; Brundage et al., 1996).
In mammalian cells, active Gαq-GTP is classically known to signal by directly binding to and activating its effector, the transmembrane enzyme phospholipase Cβ (PLCβ), which hydrolyzes the membrane lipid phosphatidylinositol 4,5 bisphosphate (PIP2) to generate the soluble molecule inositol 1,4,5 trisphosphate (IP3) and the membrane lipid diacylglycerol (DAG). IP3 can go on to increase intracellular Ca2+, which along with DAG can bind and activate protein kinase C (Figure 3; reviewed by Sánchez-Fernández et al., 2014). Prior to studies of Gαq signaling in C. elegans, the only confirmed direct Gαq effector was PLCβ. Genetic analysis of Gαq signaling in C. elegans (previously reviewed by Perez-Mansilla and Nurrish, 2009) supported the idea that PLCβ (EGL-8 in C. elegans) is a physiologically important effector (Lackner et al., 1999, Miller et al., 1999; Bastiani et al., 2003). However, these same studies also suggested that there had to be additional Gαq effector(s) since the phenotypes of PLCβ null mutants were much less severe than those of Gαq null mutants.
The missing Gαq effector was identified as the Trio RhoGEF through genetic screens for suppressors of the slow growth and hyperactive locomotion phenotypes caused by excessive Gαq pathway activity (Williams et al., 2007). Analysis using loss- and gain-of-function mutants in various double-mutant combinations showed that for control of growth, locomotion, and egg laying, Gαq signals through both PLCβ and Trio RhoGEF. Knocking out either effector alone only partially blocks Gαq signaling, while knocking out both appears to completely block Gαq signaling (Williams et al., 2007). Trio RhoGEF proved to be as or more important than the classical PLCβ effector pathway for multiple effects of Gαq signaling, including effects on growth, locomotion, and egg laying. Concomitant with the discovery of Trio RhoGEF as a Gαq effector in C. elegans, biochemical and structural studies demonstrated that mammalian Gαq binds and activates the orthologous mammalian RhoGEF proteins (Lutz et al., 2007; Rojas et al., 2007).
|
Figure 3. Mechanism of Gαq signaling. Gαq is induced to bind GTP to generate its active form by GPCRs, and induced to hydrolyze GTP to generate its inactive form by the RGS protein EAT-16. RIC-8 promotes Gαq signaling by acting as a chaperone to stabilize Gαq protein and/or by acting as a non-receptor activator to promote GTP binding by Gαq. These proteins are drawn at both left and right to facilitate showing the two Gαq effector pathways at center. Left pathway: Gαq-GTP activates Trio RhoGEF, which in turn activates sphingosine kinase (SphK) to generate the lipid second messenger sphingosine 1-phosphate (Sph1P). Trio RhoGEF also increases levels of another lipid second messenger diacylglycerol (DAG) by activating RhoA to prevent diacylglycerol kinase (DGK) from converting DAG to phosphatidic acid (PA). Right pathway: Gαq-GTP activates phospholipase Cβ (PLCβ) to generate the second messengers DAG and (not shown) IP3. Sph1P and DAG each help UNC-13 to relocalize to synaptic membranes and to promote synaptic vesicle release. DAG also activates protein kinase C (PKC) to promote dense-core vesicle release. Solid grey arrows (or bars), activation (or inhibition) via direct physical interactions. Dashed arrows or bars, indirect or poorly understood effects. Light green, proteins that promote Gαq signaling; red, proteins that inhibit Gαq signaling.
Two mechanisms for signaling downstream of the Trio RhoGEF effector of Gαq have been identified based on C. elegans genetics (Figure 3). First, while PLCβ enzyme activity directly produces DAG, Trio RhoGEF can also increase DAG levels by an indirect mechanism. Trio RhoGEF activates the RhoA protein RHO-1, allowing it to directly bind and inhibit the diacylglycerol kinase DGK-1, blocking the first step in metabolic clearance of DAG, its phosphorylation to produce phosphatidic acid (PA) (McMullan et al., 2006). Thus both effectors for Gαq collaborate to increase DAG levels. Although Rho activity in mammals is principally known to regulate other processes, such as actin cytoskeleton dynamics and transcription, C. elegans genetics demonstrates that in its role mediating neurotransmitter signaling downstream of Gαq, a major physiologically significant role of Rho seems to be increasing DAG levels.
A second downstream mediator of the effects of Gαq/RhoGEF/RhoA signaling is the sphingosine kinase SphK (SPHK-1 in C. elegans). SphK catalyzes the phosphorylation of the membrane lipid sphingosine (Sph) to sphingosine-1-phosphate (Sph1P), a signaling lipid that among other functions promotes neurotransmitter release (Okada et al., 2009). In ventral cord cholinergic neurons, Gαq signaling via the Trio RhoGEF UNC-73 causes an increase in the perisynaptic localization of SphK, and this in turn appears to increase acetylcholine release (Chan et al., 2012). In a separate study, it was found that Gαq signaling recruits SphK to synapses via calcium influx using a mechanism involving the calcium binding protein CIB (CALM-1 in C. elegans) (Chan et al., 2012). The ability of SphK to increase neurotransmitter release depends on both its localization to perisynaptic regions and its ability to generate sphingosine 1-phosphate (Chan et al., 2012). At this point, the details of the mechanism by which Trio RhoGEF leads to recruitment of SphK to perisynaptic regions, and specifically the relationship between the calcium and Trio RhoGEF effects on Sphk localization, remain to be determined.
How does the second messenger DAG produced by Gαq signaling lead to the ultimate effect of Gαq signaling, an increase in neurotransmitter release? Two DAG- binding proteins appear to mediate independent effects. One of these, UNC-13, is a protein essential for synaptic vesicle priming that facilitates neurotransmitter release by binding to the SNARE protein complexes that mediate vesicle fusion (James and Martin, 2013). Binding of UNC-13 to DAG in the membrane may help recruit it to synaptic membranes. The evidence for Gαq acting through UNC-13 includes the observations that in ventral cord motor neurons, Gαq activation of acetylcholine release depends on UNC-13, and that Gαq activation or addition of DAG analogs induces overexpressed UNC-13S::GFP to relocalize from a diffuse pattern in axons to punctate structures at synapses (Lackner et al., 1999). These effects can be abrogated by a mutation that blocks the ability of overexpressed UNC-13S::GFP to bind DAG. Recent electrophysiological studies provide perhaps the strongest evidence that UNC-13 functions in Gαq signaling to promote acetylcholine release from ventral cord motor neurons, with both the long and short isoforms of UNC-13 differentially affecting tonic and evoked release (Hu et al., 2015). Nevertheless, the extent to which UNC-13 mediates the behavioral effects of Gαq signaling has remained unclear. The recent development of methods for precise genome engineering using CRISPR/Cas9 provide an opportunity to mutate the DAG binding site of native UNC-13 and potentially resolve this issue.
A second DAG binding protein that mediates effects of Gαq signaling in ventral cord motor neurons is protein kinase C (PKC) (Sieburth et al., 2007). PKC appears to affect release of neuropeptide-containing dense-core vesicles, although the targets via which PKC does this remain unknown. PKC does not appear to directly affect release of acetylcholine from small-clear synaptic vesicles. UNC-13 appears to affect small-clear vesicles, and there are differences of opinion as to whether it directly affects dense-core vesicles release (Sieburth et al., 2007; Speese et al., 2007). Thus the current model is that Gαq signaling produces DAG to act via two different DAG-binding proteins, PKC and UNC-13, that together activate release of dense-core and synaptic vesicles (Figure 3).
A key observation about the role of DAG in Gαq signaling is that treating Gαq mutant worms with the DAG analogs known as phorbol esters rescues the paralysis of Gαq null and strong reduction-of-function mutants to wild-type levels of coordinated locomotion (Reynolds et al., 2005; Williams et al., 2007). The effectiveness of such non-localized phorbol ester treatment suggests that DAG is a “licensing factor” that promotes neurotransmitter release, i.e., that DAG need not be produced focally at synapses. Further, the fact that Gαq mutants can be rescued by phorbol ester treatment demonstrates that developmental defects do not contribute to the strong paralysis of Gαq strong loss-of-function or null mutants.
How does the other second messenger produced by Gαq signaling, sphingosine 1-phosphate, increase neurotransmitter release? An initial study suggested the effect of Gαq on neurotransmitter release via SphK appeared to be entirely independent and parallel to the ability of Gαq signaling to increase neurotransmitter release via increasing DAG levels (Chan et al., 2012). However, a subsequent study suggested SphK is essential for Gαq signaling to relocalize UNC-13S::GFP to synapses (Chan and Sieburth, 2012), and UNC-13S::GFP relocalization also appears to depend on DAG (Lackner et al., 1999). So, more work remains to clarify the relationship of DAG and SphK signaling downstream of Gα in regulating neurotransmitter release.
Two upstream regulators of Gαq were identified through C. elegans genetic analysis. The Regulator of G protein signaling (RGS) protein EAT-16 behaves as a specific inhibitor of Gαq signaling (Hajdu-Cronin et al., 1999). The RIC-8 protein promotes Gαq signaling (Miller et al., 2000). I will elaborate on studies of RGS proteins and RIC-8 in Section 3.6 and Section 3.7.
The Gαs signaling pathway (Figure 4) has been intensively studied for decades in mammalian cells using biochemical approaches (Godinho et al., 2015). Gαs activated by GPCRs binds and activates its effector, the transmembrane enzyme adenylyl cyclase, which generates the second messenger cyclic AMP (cAMP). cAMP binds the regulatory subunits of protein kinase A (PKA), causing them to dissociate from and thus activate the catalytic subunit, which can then phosphorylate various target proteins. Signaling is terminated in part through the action of the enzyme phosphodiesterase to hydrolyze cAMP. Gαs is unique among Gα isoforms in that no RGS protein is known to act on Gαs to help it hydrolyze GTP, so the slow intrinsic GTPase activity of Gαs is apparently used to terminate Gαs activity.
|
Figure 4. Mechanism of Gαs signaling. Gαs is induced to bind GTP to generate its active form by GPCRs. RIC-8 promotes signaling, either via its Gα chaperone function or as a non-receptor activator. Gαs-GTP inactivates itself (not shown) via its slow intrinsic GTPase activity. Gαs-GTP activates the transmembrane enzyme adenylyl cyclase to generate the second messenger cyclic AMP (cAMP), which ultimately is hydrolyzed by phosphodiesterase. cAMP binds the regulatory subunits of protein kinase A (PKA), causing them to release the active subunit. PKA phosphorylation of unknown target proteins activates release of both synaptic and dense-core vesicles to increase neurotransmitter release. Solid grey arrows indicate direct physical interactions. Dashed arrow indicates a poorly understood effect. Green, proteins that promote Gαs signaling; red, proteins that inhibit Gαs signaling.
Genetic studies show that the same pathway delineated by biochemical studies in mammals also operates in C. elegans motor neurons (Figure 4; Schade et al., 2005). Initial studies showed that transgenic overexpression of constitutively-active Gαs kills neurons through a cAMP-dependent mechanism, as the killing could be suppressed with mutations in the adenylyl cyclase ACY-1 (Korswagen et al., 1997; Berger et al., 1998; Moorman and Plasterk, 2002). Gαs null mutants are lethal (Korswagen et al., 1997), but mutants with increased Gαs signaling are viable, and have provided important tools for genetic analysis of Gαs signaling. Mutations that increase Gαs signaling have been identified in Gαs itself (GSA-1 in C. elegans), adenylyl cyclase (ACY-1), protein kinase A (loss of the regulatory subunit KIN-2 causes constitutive activity of the catalytic subunit KIN-1), and in phosphodiesterase (PDE-4) (Schade et al., 2005; Charlie et al., 2006b). Increased Gαs signaling results in smoothly hyperactive sinusoidal locomotion, which is distinct from the hyperactive locomotion with abnormally deep body bends induced by loss of Gαo signaling or increased Gαq signaling (Schade et al., 2005). Epistasis analysis using Gαs signaling pathway mutants supports the idea that Gαs in C. elegans functions in a pathway analogous to the mammalian Gαs signaling pathway that had been characterized biochemically (Schade et al., 2005; Reynolds et al., 2005).
Gαs pathway activity requires the guanine nucleotide exchange factor RIC-8, (Reynolds et al., 2005), although it remains unclear if this is due to the function of RIC-8 as chaperone or due to RIC-8 acting as a non-receptor nucleotide exchange protein.
The ultimate effect of Gαs signaling in neurons appears to be to promote neurotransmitter release. Schade et al. (2015) showed that activating Gαs signaling in both neurons and muscles using gain-of-function mutations appears to induce acetylcholine release from motor neurons, although Reynolds et al. (2005) showed that loss of acy-1 just in neurons did not seem to affect neurotransmitter release. Separate studies in ALA neurons show that PKA activation can induce dense-core vesicle release (Zhou et al., 2007), so Gαs signaling may be able to activate neurotransmitter release via both synaptic and dense-core vesicles. The downstream targets of phosphorylation by PKA that increase vesicle release remain unknown.
Gαs signaling may operate in muscles, in addition to neurons, to control C. elegans behavior. Null mutants for the adenylyl cyclase ACY-1 show two separable defects: larval arrest and near-paralysis. In an acy-1 null mutant, re-expression of acy-1 in either muscles or neurons can rescue the larval arrest, but neither is sufficient to rescue paralysis, indicating that acy-1 may be required in both neurons and muscles for proper locomotion (Reynolds et al., 2005). Rescue experiments also indicate that Gαs signaling is required for adult neural function, but is not required for neural development, since induced expression of acy-1 in paralyzed animals lacking neural acy-1 can restore locomotion behavior in these animals (Reynolds et al., 2005).
Genetic studies initiated to study Gαs signaling have led to a broader genetic analysis of the in vivo functions of cAMP in C. elegans. While the adenylyl cyclase ACY-1 mediates all known effects of Gαs signaling (Schade et al., 2005), there are other adenylyl cyclase isoforms in C. elegans that appear to generate a Gαs-independent pool of cAMP that also affect locomotion and larval growth (Charlie et al., 2006b). cAMP is an important negative regulator of sleep-like states throughout the animal kingdom (Zimmerman et al., 2008), and Gαs pathway activating mutants are being used to study sleep-like states in C. elegans (Belfer et al., 2013). Ghosh-Roy et al., (2010) has also used Gαs pathway mutants to study the role of cAMP in axon regeneration.
The mechanism of Gαo signaling (Figure 5) is poorly understood and remains an area ripe for further investigation using the C. elegans system. Biochemical studies in mammals have shown that Gαo is expressed throughout the nervous system and is by orders of magnitude the most abundant Gα protein in neurons, constituting ∼1.5% of membrane protein in the brain (Sternweis and Robishaw, 1984). Despite efforts to identify Gαo binding proteins that might function as effectors for Gαo or its paralogs (Chen et al., 1999; Takesono et al., 1999; Cuppen et al., 2003), no such protein has been validated as a bona fide effector of Gαo. Activation of Gαo releases Gβγ subunits, allowing them to activate specific K+ channels and inhibit specific Ca2+ channels, resulting in inhibition of neural activity (Reuveny et al., 1994; Herlitze et al., 1996).
|
Figure 5. Mechanism of Gαo signaling. Gαo is induced to bind GTP to generate its active form by GPCRs, or by the non-receptor activators AGS-3 and RIC-8. Alternatively, or in addition, RIC-8 may function as a chaperone for Gαo. Gαo-GTP may signal to inhibit the neurotransmitter release machinery by an unknown effector protein. The Gβγ subunits released from Gαo can activated specific K+ channels to reduce electrical excitability, which in turn facilitates neurotransmitter release. Green, proteins that promote Gαo signaling; red, proteins that inhibit Gαo signaling.
The ultimate effect of Gαo signaling in C. elegans neurons is to inhibit neurotransmitter release, and this has been studied in detail in ventral cord motor neurons, where Gαo signaling inhibits acetylcholine release (Nurrish et al., 1999; Miller et al., 1999; Charlie et al., 2006a; Vashlishan et al., 2008), and in HSN neurons, where Gαo signaling inhibits release of serotonin and other neurotransmitters that stimulate egg laying (Tanis et al., 2008). Gαq and Gαo signaling have the opposite effects on locomotion, egg laying, and a variety of other behaviors (Movie 1), suggesting that in general, while Gαq signaling promotes neurotransmitter release (see Section 3.2), Gαo has an opposing effect of inhibiting neurotransmitter release.
Activation of Gαq signaling in ventral cord motor neurons appears to increase the localization of the synaptic vesicle priming protein UNC-13S to presynaptic termini, as seen by visualizing a fusion of UNC-13S to the green fluorescent protein (Lackner et al., 1999). A Gαo loss-of-function mutation also causes an increase in localization of this UNC-13S::GFP protein to presynaptic termini (Nurrish et al., 1999). This result suggests that that Gαq and Gαo oppose each other at a mechanistic level. The effects of Gαq and Gαo on UNC-13S::GFP localization both depend on the diacylglycerol (DAG) binding site of UNC-13 (Lackner et al., 1999; Nurrish et al., 1999). Thus it is assumed that the increase in DAG levels produced by Gαq signaling cause the observed changes in UNC-13S::GFP localization. Gαo signaling could affect UNC-13::GFP and inhibit neurotransmitter release by decreasing DAG levels (Nurrish et al., 1999). However, no biochemical experiments have demonstrated any effect of Gαo on DAG or on the enzymes that generate or degrade DAG (Jose and Koelle, 2005; Perez-Mansilla and Nurrish, 2009).
While genetic studies in C. elegans have not clarified the downstream mechanism of Gαo signaling, they have identified novel upstream regulators of Gαo. The RGS protein EGL-10 is expressed in all neurons, and EGL-10 null mutants show sluggish locomotion and egg-laying defects due to increased Gαo signaling (Koelle and Horvitz, 1996). There are other RGS proteins (RGS-1, RGS-2, RGS-7) that additionally inhibit Gαo in specific cell types or under specific circumstances (see Section 3.7). Gαo signaling is promoted by RIC-8, working in combination with a GPR-domain containing protein (Section 3.6). As in the case of RIC-8’s effect on Gαs signaling (Section 3.3), it is unclear whether RIC-8’s effect on Gαo signaling reflects its nucleotide exchange activity, its Gα chaperone function (Gabay et al., 2011; Chan P. et al., 2013), or both.
One view is that Gαo has no effector, and that Gαo signaling occurs simply by release of Gβγ subunits that would then be entirely responsible for further downstream signaling by regulating K+ and Ca2+ channels. The plausibility of such a scheme is bolstered by careful genetic studies of heterotrimeric G protein signaling in yeast, in which mating pheromone signaling through a heterotrimeric G protein is indeed carried out by activation of a Gα protein whose principal function is simply to release Gβγ so that Gβγ can induce further signaling events (Dohlman and Thorner, 2001).
Genetic studies in C. elegans strongly suggest that Gαo signaling does not proceed purely through Gβγ effectors, and that Gαo must also signal directly through its own yet-to-be-discovered effectors. The only study of Gβγ effectors in C. elegans showed that when the EGL-6 neuropeptide receptor activates Gαo in the HSN neurons, the resulting inhibition of egg laying requires IRK-1, a homolog of mammalian Gβγ-activated K+ channels, and that no other such K+ channel is required (Emtage et al., 2012). However, this same study also demonstrated that IRK-1 does not mediate inhibition of egg laying when other methods are used to activate Gαo, suggesting that other effectors besides K+ channels must mediate Gαo signaling. This study leaves open the possibility that these other effectors could be additional Gβγ effectors such as Ca2+ channels. However, other genetic results argue in favor of effectors for Gαo itself.
First, Gαq and Gαo release the identical Gβγ subunits, as there is only one Gβγ isoform generally expressed in C. elegans neurons, yet activation of Gαq and Gαo, acting in the same neurons, have exactly opposite effects on neurotransmitter release and behavior (Lackner et al., 1999; Nurrish et al., 1999; Tanis et al., 2008). To argue that Gαo signaling principally results from released Gβγ inhibiting neural activity, one needs to explain how Gαq signaling does not give the same result. This discrepancy might result from the far greater abundance of Gαo, or by supposing that Gαo and Gαq reside in different membrane microdomains, but so far there is no actual data to support either idea.
Second, the genetics of Gαo signaling in C. elegans does not match the genetics of Gβγ-mediated pheromone signaling pathway in yeast. In the yeast pheromone signaling pathway, a null mutant for the Gα subunit results in constitutive signaling, as Gα is no longer present to sequester Gβγ and inhibit its ability to signal (Whiteway et al., 1989). Conversely, transgenic overexpression of yeast Gα inhibits pheromone signaling, apparently by increasing the pool of Gα available to associate with and inhibit Gβγ (Cole et al., 1990). The analogous experiments in C. elegans with Gαo give precisely the opposite results. Gαo null mutations appear to result in a loss of signaling, not constitutive signaling (Mendel et al., 1995; Ségalat et al., 1995), and overexpression of wild-type Gαo appears to result in constitutive signaling, not loss of signaling (Mendel et al., 1995). These results are exactly analogous to those obtained from genetic studies of Gαq in C. elegans (Brundage et al., 1996), a case in which it is clear that downstream signaling is due to Gα effectors, not Gβγ effectors.
Could it be that Gαo signals by directly binding to and inhibiting adenylyl cyclase? Adenylyl cyclase is the effector directly activated by Gαs (see Section 3.3.), and it can also be directly bound and inhibited by members of the Gαi/o family of Gα proteins, at least in cultured cells. Indeed, Gαo overexpressed in cultured cells has been shown to inhibit one isoform of adenylyl cyclase (Näsman et al., 2002). While this hypothesis is intriguing, there is currently no genetic evidence that inhibition of adenylyl cyclase is a physiologically relevant mechanism for Gαo signaling. If Gαo signaled by inhibiting adenylyl cyclase, we might expect that Gαo and Gαs signaling would show precisely opposite effects in C. elegans genetic experiments. However, this is not the case (see Section 3.3), and Gαo signaling rather shows far more precise opposition to the effects of Gαq signaling.
Could it be that Gαo signals by directly binding to and regulating one of the components of the Gαq signaling pathway, thus neatly solving the problem of identifying the Gαo effector and explaining how Gαo signaling opposes Gαq signaling (Section 3.5)? Two specific Gαq signaling pathway proteins (Figure 3) have been proposed as Gαo effectors: DGK and EAT-16. First, all genetic studies are consistent with the possibility that Gαo could signal by activating diacylglycerol kinase (DGK), the enzyme that terminates Gαq signaling by destroying the Gαq second messenger diacylglycerol. However, all efforts to demonstrate a biochemical effect of Gαo on DGK activity have proven negative (Jose and Koelle, 2005; Perez-Mansilla and Nurrish, 2009). The second hypothesis is that the effector for Gαo could be the Gαq-specific RGS protein EAT-16. An elegant model based on both genetic analysis and structural features of the EAT-16 protein complex proposes that inactive Gαo sequesters EAT-16, and activation of Gαo releases EAT-16 to inhibit Gαq (Robatzek et al., 2001). However, no biochemical studies of either the C. elegans proteins nor their mammalian equivalents has produced any support for this model.
If Gαo indeed signals directly through its own effectors, why have these effectors not yet been identified? Known effectors for other G proteins are often multi-subunit membrane protein complexes, and the cDNA expression library screening methods that have been employed in attempts to identify Gαo effectors (Chen et al., 1999; Takesono et al., 1999; Cuppen et al., 2003) are not well suited for finding such proteins. Genetic screens in C. elegans for Gαo signaling proteins would have failed to identify Gαo effectors if there are multiple redundant effectors, such that mutating a single effector gene causes little reduction in Gαo signaling. In addition, if a Gαo effector is essential for viability or reproduction, mutating such an effector would not produce animals that could be detected and recovered in a genetic screen. Because there are perfectly reasonable explanations why a Gαo effector may have eluded discovery, the absence of such a discovery at this point should not be taken as evidence that Gαo effectors do not exist.
Gαq, Gαs, and Gαo signaling all operate at the same time in the same neurons, and Figure 2 presents a model showing them acting in parallel to each other until they intersect downstream by ultimately all regulating neurotransmitter release. However, genetic epistasis experiments have been used to examine the relationship between the three G protein signaling pathways, and other relationships between the G protein signaling pathways are also consistent with these genetic results.
Double mutants for Gαq and Gαo resemble Gαq single mutants, and this and results from other double-mutant combinations are consistent with the interpretation that Gαo signaling acts to inhibit Gαq signaling (Hajdu-Cronin et al., 1999; Miller et al., 1999; Charlie et al., 2006a). More specifically, this means that Gαo signaling could inhibit the Gαq pathway at any level, upstream of Gαq, at the level of Gαq, or at any level downstream of Gαq. Only molecular experiments will ultimately be able to distinguish between these possibilities.
The relationship between Gαs and Gαq signaling has been investigated by combining mutations that increase signaling in one pathway with mutations that block signaling in the other pathway (Reynolds et al., 2005; Charlie et al., 2006a). This was done in both directions (i.e., a Gαs pathway gain-of-function mutation combined with a Gαq null mutation, and also a Gαq gain-of-function mutation combined with a Gαs pathway null mutation). The results of these experiments do not yield a simple epistatic relationship between the two signaling pathways, but do yield several important insights. First, the Gαs pathway is virtually completely dependent on the Gαq pathway to exert its effects on locomotion (Reynolds et al., 2005). Second, the Gαq pathway can still promote locomotion in the absence of a functional Gαs pathway, but the resulting locomotion is uncoordinated (Reynolds et al., 2005). Third, phorbol esters (analogs of the DAG ultimately produced by the Gαq pathway) can also promote uncoordinated locomotion in the absence of a functional Gαs pathway, suggesting Gαs signaling regulates locomotion downstream of DAG production (Reynolds et al., 2005). Fourth, the Gαq and Gαs pathways have distinct effects in driving locomotion, one line of evidence for which is that loss of Gαq signaling appears to strongly reduce acetylcholine release from motor neurons while loss of Gαs signaling does not (Charlie et al., 2006a).
An important contribution of the C. elegans system was the discovery of the receptor-independent Gα chaperone and activator RIC-8, which was first identified in a genetic screen for mutants with reduced acetylcholine release from ventral cord motor neurons (Miller et al., 1996). Genetic studies showed that reduction of function mutations in RIC-8 and Gαq have similar phenotypes and were consistent with RIC-8 acting upstream of Gαq to promote Gαq signaling (Miller et al., 2000). Subsequent genetic studies demonstrated that RIC-8 also appears to promote Gαs signaling (Reynolds et al., 2005) and Gαo signaling (Miller and Rand, 2000; Hofler and Koelle, 2011).
Biochemical studies of mammalian RIC-8 have identified two mechanisms by which RIC-8 can promote Gα signaling. First, RIC-8 can act as a nucleotide exchange factor to convert inactive Gα-GDP to active Gα-GTP (Tall et al., 2003; Chan et al., 2011). Unlike GPCRs, which are transmembrane proteins that act as nucleotide exchange factors to activate Gαβγ heterotrimers, RIC-8 is a soluble protein and will not act on the heterotrimer, but rather can only activate free Gα-GDP. This leads to the model depicted in Figure 1 in which a Gαβγ heterotrimer would need to initially be activated by a GPCR to generate Gα-GTP, but after GTP hydrolysis the inactive Gα-GDP produced might be reactivated by the nucleotide exchange activity of RIC-8, thus prolonging signaling.
The second mechanism by which RIC-8 can activate Gα signaling is by simply stabilizing Gα proteins. Knocking out RIC-8 in mammalian cells causes dramatic reductions in the levels of Gα proteins (Gabay et al., 2011). Biochemical studies show RIC-8 acts as a chaperone to help fold nascent Gα proteins (Chan P. et al., 2013). It has not yet been determined if RIC-8 mutations in C. elegans similarly cause reductions of Gα protein levels. The genetics of RIC-8 are consistent with RIC-8 promoting Gα signaling either by simply stabilizing Gα proteins, by acting as a nucleotide exchange factor, or by both mechanisms.
Another type of receptor-independent activator for Gαo are proteins containing a short motif that binds specifically to Gα proteins of the Gαi/o subfamily and that is known either as the GPR (G protein regulatory) or GoLoco motif (Siderovski et al., 1999; Peterson et al., 2000). C. elegans has three such proteins. The two very similar and functionally redundant GPR-1 and GPR-2 proteins act in early embryonic cells with Gαo to regulate spindle positioning during asymmetric cell divisions (Colombo et al., 2003; Gotta et al., 2003; Srinivasan et al., 2003). The AGS-3 protein acts with Gαo to regulate function of adult neurons (Hofler and Koelle, 2011). Biochemical studies of mammalian GPR/GoLoco proteins emphasize the fact that when these proteins bind Gα-GDP, they inhibit nucleotide exchange, which would suggest that they prevent formation of active Gα-GTP and thus inhibit signaling (Siderovski et al., 1999; Peterson et al., 2000). However, C. elegans genetic studies clearly demonstrate that precisely the opposite is true, since GPR-1/2 promote Gαo activity in embryonic cell divisions and AGS-3 also promotes Gαo signaling in adult neurons (Colombo et al., 2003; Gotta et al., 2003; Srinivasan et al., 2003; Hofler and Koelle, 2011). The apparent conflict between these results can be resolved by the findings that all known functions of the GPR/GoLoco proteins in C. elegans also require RIC-8, along with the biochemical result that RIC-8 can act as a nucleotide exchange factor on the complex between Gαo-GDP and a GPR/GoLoco protein to produce active Gαo-GTP (Thomas et al., 2008). Thus an attractive model is that when active Gαo-GTP hydrolyzes GTP to become Gαo-GDP, the function of the GPR/GoLoco protein is to bind Gαo-GDP, preventing it from reassociating with Gβγ, which would have terminated signaling, and rather to present Gαo-GDP to RIC-8 for nucleotide exchange to convert it back to active Gαo-GTP (Figure 1). The net result would be to prolong signaling that had been initiated earlier by a GPCR.
The C. elegans GPR/GoLoco protein AGS-3 is of particular interest as it has a close mammalian brain ortholog, AGS3, and a role in mediating behavioral response to food deprivation. AGS-3 is expressed in most or all C. elegans neurons and is required for several different behavioral changes that normally occur in response to short-term food deprivation (Hofler and Koelle, 2011). Biochemical studies show that over the first few hours of food deprivation, AGS-3 protein from C. elegans lysates changes from a detergent-insoluble form to a detergent-solubilizable form, indicating that a physical change in the protein occurs. These results lead to a model in which food deprivation leads to a physical change in AGS-3, allowing it to activate Gαo signaling to induce behavioral changes appropriate for the food-deprived state of the animal. It will be fascinating to see if this role of AGS-3 in mediating response to food deprivation is conserved in mammals (Hofler and Koelle, 2012).
Another important contribution of the C. elegans system was the discovery and characterization of the family of Regulator of G protein Signaling (RGS) proteins. The first RGS protein, Sst2p, was identified in yeast (Dietzel and Kurjan, 1987), but it was not clear that it had any homologs in higher eukaryotes. Genetic studies identified the C. elegans RGS protein EGL-10 as an inhibitor of Gαo signaling, and comparison of Sst2p and EGL-10 allowed the discovery of an “RGS domain” conserved between the two and that is also found in a large family of proteins in C. elegans and mammals (Koelle and Horvitz, 1996). Biochemical studies showed that the RGS domain binds directly to Gα proteins, and by stabilizing the transition state for GTP hydrolysis, inactivates signaling by driving the conversion of active Gα-GTP to the inactive species Gα-GDP (Berman et al., 1996; Tesmer et al., 1997). GTPase activators were previously known for the small ras-like G proteins, which have structural similarity to the GTPase domain of Gα proteins but lack a catalytic arginine residue that Gα proteins use for GTP hydrolysis. Thus small G proteins are unable to convert GTP to GDP at a meaningful rate without first binding a GTPase activating protein that inserts an arginine residue into their active site (Li and Zhang, 2004). In contrast, Gα subunits of heterotrimeric G proteins were initially thought to not require help from any GTPase activating proteins to convert to their inactive GDP-bound state, since they have an intrinsic GTPase activity orders of magnitude faster than that of small GTPases. However, genetic studies in C. elegans show that in vivo, the Gαo and Gαq proteins require RGS proteins to appropriately terminate signaling (Koelle and Horvitz, 1996; Hajdu-Cronin et al., 1999). In contrast, neither genetic studies in worms nor biochemical studies of mammalian proteins have identified a GTPase activator for Gαs. Thus Gαs may indeed simply use its slow intrinsic GTPase activity to terminate signaling.
The two most intensively studied RGS proteins in C. elegans are EGL-10 and EAT-16, which serve as specific inhibitors of Gαo and Gαq signaling, respectively (Koelle and Horvitz, 1996; Hajdu-Cronin et al., 1999). They are members of the R7 family of RGS proteins, of which there are four members in mammals that are expressed in the brain (Anderson et al., 2009). In vitro, these mammalian R7 proteins all preferentially inactivate Gαo rather than Gαq, and it is unclear if any of them might actually be Gαq specific in vivo, like C. elegans EAT-16. Analysis of chimeras between EGL-10 and EAT-16 shows that the Gαo versus Gαq specificity of these proteins lies in an N-terminal domain that remains of unknown biochemical function (Patikoglou and Koelle, 2002). The C. elegans R7 RGS proteins, like their mammalian homologs, contain a Gγ-like domain that is constitutively bound to a Gβ-like protein known as Gβ5 in mammals and GPB-2 in C. elegans (Chase et al., 2001; Robatzek et al., 2001; van der Linden et al., 2001). Thus the R7 RGS/Gβ complex in many respects resembles a Gβγ complex, but there is as yet no understanding of what this means or how it might be used. EAT-16, like the mammalian R7 RGS proteins, is anchored to the plasma membrane via a lipid-modified membrane anchoring subunit (Porter and Koelle, 2009). There is no known membrane anchor for EGL-10.
There are at least 13 RGS proteins in C. elegans, and the genes for all have been knocked out and studied (reviewed by Porter and Koelle, 2009). While the R7 family RGS proteins EGL-10 and EAT-16 are widely expressed, and mutants cause severe disruption of G protein signaling and behavior, the other RGS proteins appear to have much more specific functions. For example, RGS-3 is expressed only in a subset of sensory neurons, where it dampens signaling in response to strong stimuli, increasing the dynamic range for sensing these stimuli (Ferkey et al., 2007). RGS-1, RGS-2, and RGS-7 are all inhibitors of Gαo, but have much more limited functions than does the main Gαo inhibitor EGL-10. RGS-7 is expressed in early embryos where it regulates the function of Gαo in controlling asymmetric cell divisions (Hess et al., 2004). RGS-1 and RGS-2 are very similar to each other and are functionally redundant (Dong et al., 2000). Knocking out both disrupts a specific behavioral response that occurs when starved animals are re-fed. Knockouts for a number of additional RGS genes so far have shown no detectable defects, suggesting that these RGS genes, like RGS-1 and RGS-2, may have redundant and/or very specific functions that will be difficult to define.
The studies in C. elegans reviewed above show that the molecular details of the Gαq, Gαs, and Gαo signaling pathways, to the extent they are known, are the same in C. elegans versus the other invertebrate and vertebrate systems in which they have been studied, until we get down to the very most downstream outputs of these signaling pathways. The C. elegans work has been focused largely on the effects of the G proteins in ventral cord motor neurons on locomotion behavior and in the HSN neurons on egg-laying behavior. Using these two behaviors as readouts, the relevant outputs of signaling that have been identified are changes in the efficiency of the neurotransmitter release machinery. All the proteins involved in these changes are highly conserved across evolution, and functional studies in mammalian neurons support the idea that the very same mechanisms are used in mammalian brain to allow G protein signaling to modulate neurotransmitter release (Rhee et al., 2002; Wierda et al., 2007).
Studies of neural G protein signaling in more complex model organisms have focused on readouts of signaling very different from those used in C. elegans. When electrophysiological measurements have been used to measure signaling outcomes, the focus has been on the regulation of specific ion channels by Gβγ subunits (Reuveny et al., 1994; Herlitze et al., 1996), and indeed some of the effects of neural G protein signaling in C. elegans are mediated by such ion channels (Emtage et al., 2012). When effects on long-term memories have been the readout of signaling, the focus has been on the lasting effects of neural G protein signaling on synaptic structure or on transcription (Kandel, 2004). It is not yet fully clear whether neural G protein signaling in C. elegans results in these sorts of lasting changes. We do know that Gαq signaling in worms, as in mammals, results in activation of the Rho GTPase, whose best-studied function is as a regulator of the actin cytoskeleton, but it remains to be seen if Gαq signaling through Rho leads to functionally significant and lasting structural changes in C. elegans synapses. In Aplysia and mammals, Gαs signaling produces cAMP that activates the transcription factor CREB to induce changes in transcription that may underlie long-term memories (Kida et al., 2002; Kandel, 2012), although the relevance of CREB is less clear in Drosophila memory (Perazzona et al., 2004). C. elegans also has a CREB homolog that was shown to be essential for worms to form a long-term memory of a food-odorant association (Kauffman et al., 2010) and that mediates G protein regulation of gene expression (Suo et al,. 2006). Thus it seems likely that the mechanisms of neural G protein signaling are deeply conserved across evolution, but that the different types of behavioral readouts used to study such signaling in different model organisms tends to focus attention on different downstream effects of the signaling pathways on neural function.
Finally, I return to the simplified model presented in Figure 2 to illustrate an overview of what has been learned from studies of neurotransmitter signaling through heterotrimeric G proteins in C. elegans. As described in Section 2, a typical neuron expresses many GPCRs, and these receptors sense the levels of neurotransmitters that are released, not just from presynaptic partners to our typical neuron, but also from distant neurons and are thus acting extrasynaptically. Each neurotransmitter and its receptor are expressed in very specific and small sets of neurons, and the extrasynaptic communication between these cells is a crucial type of functional connection that goes beyond the physical synaptic connections between neurons and that is essential for understanding neural circuits. As described in Section 3, three different Gα proteins, Gαq, Gαs, and Gαo, integrate signaling by the many GPCRs that may be active on a neuron at any one time. The major outcomes of neural G protein signaling studied in C. elegans are the opposing effects that Gαq and Gαo have to activate and inhibit the synaptic vesicle release machinery, respectively. Gαs signaling collaborates with the Gαq pathway to promote synaptic output. Other outcomes of Gα activation, such as regulation of K+ and Ca2+ channels, or effects on neural structure or transcription, also seem to occur but are not responsible for the most obvious effects of the G proteins on C. elegans behavior. The neurotransmitters, receptors, G proteins, downstream signaling molecules, and mechanisms used in C. elegans are all conserved in mammals. The contribution of C. elegans studies has been to focus attention on the ubiquity of extrasynaptic signaling through GPCRs, the effects of neural G proteins on the neurotransmitter release machinery, and on new signaling molecules such as RIC-8, TrioRhoGEF, and RGS proteins that were discovered in C. elegans using the power of forward genetic screens.
Opportunities for future progress using C. elegans include the chance to grapple with the huge family of GPCRs that activate neural G proteins. Perhaps the entire set of GPCRs expressed on some model neurons, such as the ventral cord motor neurons or the HSN egg-laying neurons, can be defined and used to study how these receptors act together to regulate activity of these neurons. More work remains to define the mechanisms by which the three major neural G proteins affect neurotransmitter release, and at what level they intersect. In particular, the effector for Gαo, if it exists, still remains to be identified. Finally, it remains to be seen whether C. elegans can be used to successfully study the types of changes in synaptic structure and transcription that occur downstream of G proteins in neurons of more complex organisms.
I thank Judy Pepper and Jacob Brewer for preparing the video shown as Movie 1, Robert Fernandez for providing research for Table 1 and Table 2, and Seongseop Kim, Robert Fernandez, and Andrew Olson for critically reading the manuscript. Work in the Koelle lab is supported by grants R01NS036918 and R01NS086932 from the NIH.
Table 1. G protein coupling and pharmacological profiles of C. elegans G protein coupled small molecule neurotransmitter receptors
| Serotonin receptors | Experimental evidence for ligand assignment | G protein coupling | References G protein coupling | References binding studies | Agonists (EC50) in heterologous cells | References heterologous cell studies |
|---|---|---|---|---|---|---|
| SER-1 | Binding studies, heterologous cell signaling, mutant resistant to serotonin | Gαq in heterologous cells | Hamdan et al., 1999 | Hamdan et al., 1999 | Serotonin (0.72 and 0.8 μM), lisuride (partial, 0.2 and 2.4 μM), DOI (partial, >100 μM). No activity with dopamine or octopamine. | Hamdan et al., 1999 |
| SER-4 | Binding studies, heterologous cell signaling, mutant resistant to serotonin | Gαi/o in heterologous cells | Olde and McCombie, 1997 | Olde and McCombie, 1997 | Serotonin (~0.1 μM), tyramine | Olde and McCombie, 1997; Petrascheck et al., 2007 |
| SER-5 | Mutant resistant to serotonin | Gαs predicted from sequence analysis | Harris et al., 2009 | |||
| SER-7 | Binding studies, heterologous cell signaling, mutant resistant to serotonin | Gαs in heterologous cells, Gαs in MC neurons, Gα12 in M4 neuron | Hobson et al., 2003; Hobson et al., 2006; Song and Avery, 2012 | Hobson et al., 2003; Hobson et al., 2006 | Serotonin (30 nM), tryptamine (4.7 μM) | Hobson et al., 2003; Hobson et al., 2006 |
| Dopamine receptors | Experimental evidence for ligand assignment | G protein coupling | References G protein coupling | References binding studies | Agonists (EC50) in heterologous cells | References heterologous cell studies |
| DOP-1 | Binding studies, mutation affects dopamine response | Gαq in ventral cord motor neurons and touch response neurons | Chase et al., 2004; Kindt et al., 2007 | Suo et al., 2002 | ||
| DOP-2 | Binding studies, mutant resistant to dopamine, heterologous cell signaling | Gαi/o in heterologous cells, GOA-1 in SIA neurons, GPA-14 in yeast interaction assay and ADE neurons. GOA-1 and GPA-7 in male mating. | Suo et al., 2003; Suo et al., 2009; Correa et al., 2012; Pandey and Harbinder, 2012; Mersha et al, 2013 | Suo et al., 2003 | dopamine (75 nM) | Suo et al., 2003 |
| DOP-3 | Mutant resistant to dopamine, heterologous cell signaling | Gαo in ventral cord motor neurons, Gαi/o in heterologous cells | Chase et al., 2004; Sugiura et al., 2005 | dopamine (27 nM), tyramine (500 nM ), octopamine (250 μM) | Sugiura et al., 2005 | |
| DOP-4 | Mutant resistant to dopamine, heterologous cell signaling | Gαs in heterologous cells | Sugiura et al., 2005 | dopamine (9.3 μM), >1 mM for dopamine, octopamine, tyramine and serotonin | Sugiura et al., 2005 | |
| Octopamine receptors | Experimental evidence for ligand assignment | G protein coupling | References G protein coupling | References binding studies | Agonists (EC50) in heterologous cells | References heterologous cell studies |
| OCTR-1 | Binding studies, heterologous cell signaling, mutant resistant to octopamine | Gαo in ASH neurons, Gαo in heterologous cells | Harris et al., 2010 | Wragg et al., 2007 | octopamine (0.39 μM in Xenopus oocytes, but binding studies suggest higher affinity) | Mills et al., 2012 |
| SER-3 | Mutant resistant to octopamine, heterologous cell signaling | Gαq in ASH neurons, Gαq in heterologous cells | Suo et al., 2006 | octopamine (24 nM in mammalian cells; 0.33 μM in Xenopus oocytes), tyramine (26 μM in mammalian cells) | Petrascheck et al., 2007; Mills et al., 2012 | |
| SER-6 | Mutant resistant to octopamine | Gαs in ADL neurons | Mills et al., 2012; | octopamine (2 μM in Xenopus oocytes) > tyramine. No response to dopamine or serotonin. | Mills et al., 2012; | |
| Tyramine receptors | Experimental evidence for ligand assignment | G protein coupling | References G protein coupling | References binding studies | Agonists (EC50) in heterologous cells | References heterologous cell studies |
| SER-2 | heterologous cell signaling, mutant resistant to tyramine | Gαi/o in heterologous cells, Gαo in VD neurons | Rex and Komuniecki, 2002; Donelly et al., 2013 | Rex and Komuniecki, 2002 | Tyramine 360 nM, 403 nM) | Rex and Komuniecki, 2002; Rex et al., 2004 |
| TYRA-2 | Binding studies, heterologous cell signaling | Gαi/o in heterologous cells | Rex et al., 2005 | Rex et al., 2005 | Tyramine (50 nM), octopamine and dopamine had little/no effect | Rex et al., 2005 |
| TYRA-3 | Binding studies, mutant resistant to tyramine | Gαq in heterologous cells | Hapiak et al., 2013 | Wragg et al., 2007 | tyramine (70.8 nM) | Hapiak et al., 2013 |
| Acetylcholine receptors | Experimental evidence for ligand assignment | G protein coupling | References G protein coupling | References binding studies | Agonists (EC50) in heterologous cells | References heterologous cell studies |
| GAR-1 | heterologous cell signaling | Gαi in Xenopus oocytes | Park et al., 2000 | Acetylcholine (~100 nM), carbachol, no effect of oxotremorine | Park et al., 2000 | |
| GAR-2 | heterologous cell signailng | Gαi/o in Xenopus oocytes, Gαo in ventral chord cholinergic neurons | Lee et al., 2000; Dittman and Kaplan, 2008 | acetylcholine (~35 nM), carbachol, no effect of oxotremorine | Lee et al., 2000; Suh et al., 2001 | |
| GAR-3 | heterologous cell signaling, mutant resistant to acetylcholine agonist arecoline | Gαq and Gαi/o in heterologous cells, Gαq in pharyngeal muscles, Gαq in male spicule protraction | Min et al., 2000; Park et al., 2003; Steger and Avery 2004; Park et al., 2006; Liu et al., 2007 | carbachol (13.0 and 11.5 μM), oxotremorine | Hwang et al., 1999; Min et al., 2000; Park et al., 2003 | |
| GABA receptors | Experimental evidence for ligand assignment | G protein coupling | References G protein coupling | References binding studies | Agonists (EC50) in heterologous cells | References heterologous cell studies |
| GBB-1 | Mutant insensitive to optogenetically released GABA | Gαo in cholinergic motor neurons | Dittman and Kaplan, 2008 | |||
| GBB-2 | Mutant insensitive to optogenet-ically released GABA | Gαo in cholinergic motor neurons | Dittman and Kaplan, 2008 | |||
| Glutamate receptor homologs | Experimental evidence for ligand assignment | G protein coupling | References G protein coupling | References binding studies | Agonists (EC50) in heterologous cells | References heterologous cell studies |
| MGL-1 | Mutant resistant to mGluR agonists trans-ACPD and LCCG-I | |||||
| MGL-2 | heterologous cell signaling | Gαq in heterologous cells | Tharmalingam et al., 2012 | glutamate (8.61 μM), quisqualate (169 μM), DHPG (partial agonist), | ||
| MGL-3 | None—inferred to be a glutamate receptor from sequence homology | |||||
| Poorly-studied putative small-molecule GPCRs | Experimental evidence for ligand assignment | G protein coupling | References G protein coupling | References binding studies | Agonists (EC50) in heterologous cells | References heterologous cell studies |
| T02E9.3 | ||||||
| C24A8.1 | ||||||
| C24A8.6 | ||||||
| F35H10.10 | ||||||
| F59D12.1 | ||||||
| T21B4.4 |
Table 2. Genetically-established functions and expression patterns of C. elegans G protein coupled small molecule neurotransmitter receptors
| Serotonin receptors | Functions revealed in mutants | References mutant phenotype | Expression pattern | References expression pattern |
|---|---|---|---|---|
| SER-1 | Exogenous serotonin-stimulated egg laying, feeding, and slowing of locomotion, α-methyl-5-HT stimulated egg laying, male ventral tail curling, food-induced slowing, food modulation of aversive response, heat shock response, longevity | Carnell et al., 2005; Dempsey et al., 2005; Xiao et al., 2006; Dernovici et al., 2007; Murakami and Murakami, 2007; Srinivasan et al., 2008; Harris et al., 2011 | Pharyngeal muscle (pm3, pm4, pm5, pm6, pm7, pm8), CEP, RMG, RMH, RMF, RMD, RIA, RIC, URY, additional head neurons, vulval muscle and epithelial cells, tail neurons (PVT, PVQ, possibly DVC) ventral nerve cord, excretory cell, uterine cells, ray sensory neurons in male | Cho et al., 2000; Tsalik et al., 2003; Carnell et al., 2005; Dempsey et al., 2005; Xiao et al., 2006; Dernovici et al., 2007 |
| SER-4 | Egg laying induced by imipramine but not by fluoxetine or serotonin, inhibition of locomotion by serotonin, effects of ethanol on gustatory plasticity and locomotion, thermotaxis memory behavior | Cho et al., 2000; Dempsey et al., 2005; Murakami and Murakami, 2007; Wang et al., 2011; Gürel et al., 2012; Li et al., 2013 | NSM, RIB or AIB, RIS, pharyngeal neuron, pair of sublateral interneurons or motorneurons, retrovesicular ganglion, PVT tail neuron, vm2 vulval muscles, DVA or DVC tail interneuron | Tsalik et al., 2003; Gürel et al., 2012 |
| SER-5 | Exogenous serotonin-stimulated egg laying, food and serotonin-dependent increase in sensitivity of ASH neurons to octanol and decrease in sensitivity to Cu2+. Reduced sensitivity to serotonin and fluoxetine induced paralysis. | Hapiak et al., 2009; Kullyev et al., 2010; Harris et al., 2011; Guo et al., 2015 | Neurons including AWB and ASH, body wall muscles, vulval muscles | Hapiak et al., 2009 |
| SER-7 | Exogenous serotonin-stimulated egg laying, pharyngeal pumping, and food intake. Regulation of pharyngeal pumping. Egg laying. Hypoxia regulation of gustatory and effects of ethanol on gustatory plasticity, thermotaxis memory behavior | Hobson et al., 2006; Donohoe et al., 2009; Hapiak et al., 2009; Pocock and Hobert, 2010; Wang et al., 2011; Song and Avery, 2012; Song et al., 2013; Li et al., 2013; Gomez-Amaro et al., 2015; Leiser et al., 2015 | Pharyngeal neurons MC, M4, I2, I3, M5, M3, I4, I6, and M2, vulval muscles | Hobson et al., 2006 |
| Dopamine receptors | Functions revealed in mutants | References mutant phenotype | Expression pattern | References expression pattern |
| DOP-1 | Antagonizes DOP-3 to control locomotion, a behavioral decision-making paradigm, and acetylcholine release. Regulates swim-to-crawl transition, fat reservoirs, local food search behavior. Delays habituation to mechanical stimulation. Promotes nicotine approach behavior. | Chase et al., 2004; Sanyal et al., 2004; Kindt et al., 2007; Allen et al., 2011; Vidal-Gadea et al., 2011; Sellings et al., 2013; Barros et al., 2014; Bhattacharya et al., 2014; Wang et al., 2014 | DVA, PLM, PHC, ALN, ALM, AVM, PLN, PVQ, AUA, RIB, RIM, RIS, unidentified head neurons, head muscles, labial and amphid sheath/socket cells, excretory gland cells, cholinergic ventral cord motor neurons | Tsalik et al., 2003; Sanyal et al., 2004; Chase et al., 2004; Bhattacharya et al., 2014 |
| DOP-2 | Antagonizes octopamine signaling, inhibits CRE-mediated gene expression in SIA. Mediates swimming-induced paralysis. Inhibits unproductive male mating behavior. Promotes nicotine approach behavior. Regulates touch habituation and chemosensory associative learning. Affects a decision-making paradigm. | Suo et al., 2009; Carvelli et al., 2010; Correa et al., 2012; Sellings et al., 2013; Mersha et al. 2013; Wang et al., 2014; Correa et al., 2015 | All dopaminergic neurons of hermaphrodite (CEP, ADE, PDE) and probably of male (R5A, R7A, R9A). RIA, SIA, SIB, RID, PDA, HOA. | Suo et al., 2003; Tsalik et al., 2003; Suo et al., 2009; Correa et al., 2015 |
| DOP-3 | Antagonizes DOP-1 to control locomotion, a behavioral decision-making paradigm, and acetylcholine release. Mediates swimming induced paralysis. Inhibits unproductive male mating behavior. Antagonizes octopamine signaling and inhibits CRE-mediated gene expression in SIA. Regulates avoidance of octanol and 2-nonanone. | Chase et al., 2004; McDonald et al., 2007; Suo et al., 2009; Ezak and Ferkey, 2010; Carvelli et al., 2010; Kimura et al., 2010; Omura et al., 2012; Correa et al., 2012; Wang et al., 2014 | Ventral cord GABAergic and cholinergic neurons, ASK, PVD, SIA, RIC, head neurons, tail neurons, body wall muscles, male tail cells | Chase et al., 2004; Suo et al., 2009; Ezak and Ferkey, 2010; Correa et al., 2012; |
| DOP-4 | Mediates swimming induced paralysis, swim/crawl transition, food enhancement of ASH response to repellents, alcohol-induced disinhibition of crawling. | Carvelli et al., 2010; Ezcurra et al., 2011; Topper et al., 2014; Vidal-Gadea et al., 2011 | I1, I2, ASG, AVL, CAN, PQR, vulva, intestine, rectal glands, rectal epithelial cells, male ray 8, additional head neurons, ASH (inferred indirectly). | Sugiura et al., 2005; Ezcurra et al., 2011 |
| Octopamine receptors | Functions revealed in mutants | References mutant phenotype | Expression pattern | References expression pattern |
| OCTR-1 | Mediates effect of exogenous octopamine on response to octanol. Controls innate immunity by regulating the unfolded protein response. | Wragg et al., 2007; Harris et al., 2010; Sun et al., 2011; Mills et al., 2012; Sun et al., 2012 | ASI, ASH, AIY, ADE, CEP, spermatheca, uterine toroid cells, head and tail neurons | Wragg et al., 2007 |
| SER-3 | Mediates octopamine- and starvation-induced expression of a CREB-dependent reporter gene in SIA neurons. Antagonizes OCTR-1 in the control of octanol response by exogenous octopamine. Mediates inhibition of ASI sensory neuron by ASH sensory neuron via a circuit involving octopamine released from RIC. | Suo et al., 2006; Mills et al., 2012; Guo et al., 2015 | ASH, ASG, ASI, SIA, PHA, PHB, PVQ, neurons in the head and tail, head muscles, intestine, phasmid socket cells, spermatheca | Suo et al., 2006; Mills et al., 2012; Guo et al., 2015 |
| SER-6 | Mediates octopamine- and starvation-induced expression of a CREB-dependent reporter gene in SIA neurons. Mediates exogenous serotonin-stimulated fat reduction. Mediates exogenous octopamine mediated inhibition of response to 100% octanol. | Srinivasan et al., 2008; Mills et al., 2012; Yoshida et al., 2014 | SIA, RIC, AWB, ADL, ASI, head neurons, tail neurons, posterior ventral cord neurons, intestine | Srinivasan et al., 2008; Yoshida et al., 2014 |
| Tyramine receptors | Functions revealed in mutants | References mutant phenotype | Expression pattern | References expression pattern |
| SER-2 | Mediates paralysis and suppression of pharyngeal pumping by exogenous tyramine. Suppresses head movements during backing. Facilitates execution of omega turns. | Rex et al., 2004; Donelly et al., 2013 | Neurons: AIY, AVH, AUA, RIC, SAB, RID, RIA, SDQ, CAN, DA9, LUA, ALN, PVC, NSM, AIZ, DVA, BDU, SIA, RME, PVT, OLL, PVD, VD. Excretory gland cells. Muscles: pm1, pm6, head muscles, male posterior body wall muscles and diagonal muscles. Possibly male CP neurons. Uterine toroid cells ut1 and ut2. | Tsalik et al., 2003; Rex et al., 2004; Donelly et al., 2013 |
| TYRA-2 | MCL, NSM, ASE, ASG, ASH, ASI, PVD, CAN, ALM | Rex et al., 2005 | ||
| TYRA-3 | Mediates effect of exogenous tyramine on response to octanol; affects decision to leave bacterial lawn. Antagonizes the effect of serotonin to sensitize aversive responses. | Wragg et al., 2007; Bendesky et al., 2011; Hapiak et a., 2013; | ADE, CEP, ASK, ADL, AIM, AUA, BAG, DEP, OLQ, SDQL, AFD, AWC, RIC, ASI, spermatheca, distal tip cell, vulval muscles, additional head and tail neurons | Carre-Pierrat et al., 2006; Unpublished results cited in Wragg et al., 2007; Bendesky et al., 2011 |
| Acetyl-choline receptors | Functions revealed in mutants | References mutant phenotype | Expression pattern | References expression pattern |
| GAR-1 | head neurons, PVM | Lee et al., 2000 | ||
| GAR-2 | Mediates inhibition of egg laying. Mediates feedback inhibition of cholinergic ventral cord motor neurons to regulate locomotion. | Bany et al., 2003; Dittman and Kaplan, 2008 | head neurons, ventral cord GABAergic and cholinergic, neurons, cell near vulva | Lee et al., 2000; Dittman and Kaplan, 2008 |
| GAR-3 | Regulates multiple calcium-dependent processes in pharyngeal muscle. Sensitizes nicotinic signaling in male spicule neurons and muscles to facilitate mating. Regulates head movements by compartmentalizing axon activity in the RIA interneuron. Promotes acetylcholine release from cholinergic motor neurons. | Steger and Avery, 2004; Liu et al., 2007; Hendricks et al., 2012; Chan J.P. et al., 2013 | I3, pharyngeal muscle, body wall muscles, anal depressor muscle, cholinergic ventral cord neurons, VD, DD, male diagonal muscles, male ray 8 neuron, SPC, PCB, and PCA neurons and the spicule protractor muscles | Steger and Avery, 2004; ; Liu et al., 2007; Chan J.P. et al., 2013 |
| GABA receptors | Functions revealed in mutants | References mutant phenotype | Expression pattern | References expression pattern |
| GBB-1 | With GBB-2, decreases acetylcholine release from ventral cord motor neurons to regulate locomotion. Affects lifespan. | Dittman and Kaplan, 2008; Schultheis et al., 2011; Chun et al., 2015 | Expression in ventral cord cholinergic neurons in inferred indirectly. | Dittman and Kaplan, 2008; Schultheis et al., 2011 |
| GBB-2 | With GBB-1, decreases acetylcholine release from ventral cord motor neurons to regulate locomotion. | Dittman and Kaplan 2008; Schultheis et al., 2011 | Expression in ventral cord cholinergic neurons in inferred indirectly. | Dittman and Kaplan 2008; Schultheis et al., 2011 |
| Glutamate receptor homologs | Functions revealed in mutants | References mutant phenotype | Expression pattern | References expression pattern |
| MGL-1 | Regulates starvation response, perhaps by directly sensing amino acids from food. Pharmacological activation of MGL-1 inhibits pharyngeal pumping. Mediates reduction in pharyngeal pumping when animals are removed from food. | Kang and Avery, 2009; Dillon et al,. 2015 | Expression in AIY and AIB is inferred indirectly. Head neurons, tail neurons, pharyngeal neurons including NSM. | Kang and Avery, 2009; Dillon et al,. 2015 |
| MGL-2 | Regulates starvation response, perhaps by directly sensing amino acids from food. | Kang and Avery, 2009 | Expression in AIY and AIB is inferred indirectly. Head neurons, tail neurons, pharyngeal neurons including NSM, I5, intestine, pharyngeal-intestinal valve, pharyngeal muscle. | Kang and Avery, 2009; Dillon et al,. 2015 |
| MGL-3 | Head neurons, pharyngeal neurons including NSM. | Dillon et al., 2015 |
Abbott, N.J., Williamson, R., and Maddock, L. (1995). Cephalopod neurobiology: neuroscience studies in squid, octopus and cuttlefish. (Oxford: Oxford University Press). Abstract
Agnati, L.F., Fuxe, K., Zoli, M., Ozini, I., Toffano, G., and Ferraguti, F. (1986). A correlation analysis of the regional distribution of central enkephalin and β-endorphin immunoreactive terminals and of opiate receptors in adult and old male rats. Evidence for the existence of two main types of communication in the central nervous system: the volume transmission and the wiring transmission. Acta Physiol. Scand. 128, 201-207. Abstract Article
Agnati, L.F., Guidolin, D., Guescini, M., Genedani, S., and Fuxe, K. (2010). Understanding wiring and volume transmission. Brain Res. Rev. 64, 137-159. Abstract Article
Allen, A.T., Maher, K.N., Wani, K.A., Betts, K.E., and Chase, D.L. (2011). Coexpressed D1- and D2-like dopamine receptors antagonistically modulate acetylcholine release in Caenorhabditis elegans. Genetics 188, 579-590. Abstract Article
Altun, Z.F. (2011). Neurotransmitter receptors in C. elegans. In WormAtlas. doi:10.3908/wormatlas.5.202 Article
Anderson, G.R., Posokhova, E., and Martemyanov, K.A. (2009). The R7 RGS protein family: multi-subunit regulators of neuronal G protein signaling. Cell Biochem. Biophys. 54, 33-46. Abstract Article
Bailey, C.H., and Kandel ER. (2008). Synaptic remodeling, synaptic growth and the storage of long-term memory in Aplysia. Prog. Brain Res. 169, 179-198. Abstract Article
Bany, I.A., Dong, M.Q., and Koelle, M.R. (2003). Genetic and cellular basis for acetylcholine inhibition of Caenorhabditis elegans egg-laying behavior. J. Neurosci. 23, 8060-8069. Abstract Article
Barberis, A., Petrini, E.M., and Mozrzymas, J.W. (2011). Impact of synaptic neurotransmitter concentration time course on the kinetics and pharmacological modulation of inhibitory synaptic currents. Front. Cell. Neurosci. 5, 6. Abstract Article
Barros, A.G., Bridi, J.C., de Souza, B.R., de Castro Júnior, C., de Lima Torres, K.C., Malard, L., Jorio, A., de Miranda, D.M., Ashrafi, K., and Romano-Silva, M.A. (2014). Dopamine signaling regulates fat content through β-oxidation in Caenorhabditis elegans. PLoS One 9, e85874. Abstract Article
Bastiani, C.A., Gharib, S., Simon, M.I., and Sternberg, P.W. (2003). Caenorhabditis elegans Gαq regulates egg-laying behavior via a PLCβ-independent and serotonin-dependent signaling pathway and likely functions both in the nervous system and in muscle. Genetics 165, 1805-1822.
Bastiani, C. and Mendel, J. Heterotrimeric G proteins in C. elegans (October 13, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.75.1, http://www.wormbook.org. Abstract Article
Beaulieu, J.M., and Gainetdinov, R.R. (2011). The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 63, 182-217. Abstract Article
Belfer, S.J,, Chuang, H.S., Freedman, B.L., Yuan, J., Norton, M., Bau, H.H., and Raizen, D,M. (2013). Caenorhabditis-in-drop array for monitoring C. elegans quiescent behavior. Sleep 36, 689-698G. Abstract Article
Bendesky, A., Tsunozaki, M., Rockman, M.V., Kruglyak, L., and Bargmann, C.I. (2011). Catecholamine receptor polymorphisms affect decision-making in C. elegans. Nature 472, 313-318. Abstract Article
Berger, A.J., Hart, A.C., and Kaplan, J.M. (1998). Gαs-induced neurodegeneration in Caenorhabditis elegans. J. Neurosci. 18, 2871-2880. Abstract Article
Berman, D.M., Kozasa, T., and Gilman, A.G. (1996). The GTPase-activating protein RGS4 stabilizes the transition state for nucleotide hydrolysis. J. Biol. Chem. 271, 27209-27212. Abstract Article
Bhattacharya, R., Touroutine, D., Barbagallo, B., Climer, J., Lambert, C.M., Clark, C.M., Alkema, M.J., and Francis, M.M. (2014). A conserved dopamine-cholecystokinin signaling pathway shapes context-dependent Caenorhabditis elegans behavior. PLoS Genet. 10, e1004584. Abstract Article
Brandt, D.R., and Ross, E.M. (1986). Catecholamine-stimulated GTPase cycle. Multiple sites of regulation by β-adrenergic receptor and Mg2+ studied in reconstituted receptor-Gs vesicles. J. Biol. Chem. 261, 1656-1664. Abstract Article
Brundage, L., Avery, L., Katz, A., Kim, U.J., Mendel, J.E., Sternberg, P.W., and Simon, M.I. (1996). Mutations in a C. elegans Gqα gene disrupt movement, egg laying, and viability. Neuron 6, 999-1009. Abstract Article
C. elegans Deletion Mutant Consortium. (2012). Large-scale screening for targeted knockouts in the Caenorhabditis elegans genome. G3 2, 1415-1425. Abstract Article
Carnell, L., Illi, J., Hong, S.W., and McIntire, S.L. (2005). The G-protein-coupled serotonin receptor SER-1 regulates egg laying and male mating behaviors in Caenorhabditis elegans. J. Neurosci. 25, 10671-1081. Abstract Article
Carre-Pierrat, M., Baillie, D., Johnsen, R., Hyde, R., Hart, A., Granger, L., and Ségalat, L. (2006). Characterization of the Caenorhabditis elegans G protein-coupled serotonin receptors. Invert. Neurosci. 6, 189-205. Abstract Article
Carvelli, L., Matthies, D.S., and Galli, A. (2010). Molecular mechanisms of amphetamine actions in Caenorhabditis elegans. Mol. Pharmacol. 78, 151-156. Abstract Article
Chan, J.P., Hu, Z., and Sieburth, D. (2012). Recruitment of sphingosine kinase to presynaptic terminals by a conserved muscarinic signaling pathway promotes neurotransmitter release. Genes Dev. 26, 1070-1085. Abstract Article
Chan, J.P., Staab, T.A., Wang, H., Mazzasette, C., Butte, Z., and Sieburth, D. (2013). Extrasynaptic muscarinic acetylcholine receptors on neuronal cell bodies regulate presynaptic function in Caenorhabditis elegans. J. Neurosci. 33, 14146-14159. Abstract Article
Chan, P., Gabay, M., Wright, F.A., and Tall, G.G. (2011). Ric-8B is a GTP-dependent G protein αs guanine nucleotide exchange factor. J. Biol. Chem. 286, 19932-19942. Abstract Article
Chan, P., Thomas, C.J., Sprang, S.R., and Tall, G.G. (2013). Molecular chaperoning function of Ric-8 is to fold nascent heterotrimeric G protein α subunits. Proc. Natl. Acad. Sci. U. S. A. 110, 3794-3799. Abstract Article
Charlie, N.K., Schade, M.A., Thomure, A.M., and Miller, K.G. (2006a). Presynaptic UNC-31 (CAPS) is required to activate the Gαs pathway of the Caenorhabditis elegans synaptic signaling network. Genetics 172, 943-961. Abstract Article
Charlie, N.K., Thomure, A.M., Schade, M.A., and Miller, K.G. (2006b). The Dunce cAMP phosphodiesterase PDE-4 negatively regulates Gαs-dependent and Gαs-independent cAMP pools in the Caenorhabditis elegans synaptic signaling network. Genetics 173, 111-130. Abstract Article
Chase, D.L. and Koelle, M.R. (2004). Genetic analysis of RGS protein function in Caenorhabditis elegans. Methods Enzymol. 389, 305-320. Abstract Article
Chase, D.L. and Koelle, M.R. Biogenic amine neurotransmitters in C. elegans (February 20, 2007), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.132.1, http://www.wormbook.org. Abstract Article
Chase, D.L., Patikoglou, G.A., and Koelle, M.R. (2001). Two RGS proteins that inhibit Gαo and Gαq signaling in C. elegans neurons require a Gβ5-like subunit for function. Curr. Biol. 11, 222-231. Abstract Article
Chase, D.L., Pepper, J.S., and Koelle, M.R. (2004). Mechanism of extrasynaptic dopamine signaling in Caenorhabditis elegans. Nat. Neurosci. 7, 1096-1103. Abstract Article
Chen, C., and Regehr, W.G. (1997). The mechanism of cAMP-mediated enhancement at a cerebellar synapse. J. Neurosci. 17, 8687-8694. Abstract Article
Chen, L.T., Gilman, A.G., and Kozasa. T. (1999). A candidate target for G protein action in brain. J. Biol. Chem. 274, 26931-26938. Abstract Article
Cheong, M.C., Artyukhin, A.B., You, Y.J., and Avery, L. (2015). An opioid-like system regulating feeding behavior in C. elegans. eLife 4, doi: 10.7554/eLife.06683. Abstract Article
Cho, J.H., Bandyopadhyay, J., Lee, J., Park, C.S., and Ahnn, J. (2000). Two isoforms of sarco/endoplasmic reticulum calcium ATPase (SERCA) are essential in Caenorhabditis elegans. Gene 261, 211-219. Abstract Article
Choi, S., Chatzigeorgiou, M., Taylor, K.P., Schafer, W.R., and Kaplan, J.M. (2013). Analysis of NPR-1 reveals a circuit mechanism for behavioral quiescence in C. elegans. Neuron 78, 869-880. Abstract Article
Chun, L., Gong, J., Yuan, F., Zhang, B., Liu, H., Zheng, T., Yu, T., Xu, X.Z., and Liu, J. (2015). Metabotropic GABA signalling modulates longevity in C. elegans. Nat. Commun. 6, 8828. Abstract Article
Coates, J.C., and de Bono, M. (2002). Antagonistic pathways in neurons exposed to body fluid regulate social feeding in Caenorhabditis elegans. Nature 419, 925-929. Abstract Article
Cohen, M., Reale, V., Olofsson, B., Knights, A., Evans, P., and de Bono, M. (2009). Coordinated regulation of foraging and metabolism in C. elegans by RFamide neuropeptide signaling. Cell Metab. 9, 375-385. Abstract Article
Cole, G.M., Stone, D.E., and Reed, S.I. (1990). Stoichiometry of G protein subunits affects the Saccharomyces cerevisiae mating pheromone signal transduction pathway. Mol. Cell. Biol. 10, 510-517. Abstract Article
Coleman, D.E., Berghuis, A.M., Lee, E., Linder, M.E., Gilman, A.G., and Sprang, S.R. (1994). Structures of active conformations of Giα1 and the mechanism of GTP hydrolysis. Science 265, 1405-1412. Abstract Article
Collins, K.M., and Koelle, M.R. (2013). Postsynaptic ERG potassium channels limit muscle excitability to allow distinct egg-laying behavior states in Caenorhabditis elegans. J. Neurosci. 33, 761-775. Abstract Article
Colombo, K., Grill, S.W., Kimple, R.J., Willard, F.S., Siderovski, D.P., and Gönczy, P. (2003). Translation of polarity cues into asymmetric spindle positioning in Caenorhabditis elegans embryos. Science 300, 1957-1961. Abstract Article
Conn, P.J., and Roth, B.L. (2008). Opportunities and challenges of psychiatric drug discovery: roles for scientists in academic, industry, and government settings. Neuropsychopharmacology 33, 2048-2060. Abstract Article
Cori, C.F., and Cori, G.T. (1947). Polysaccharide Phosphorylase. In Nobel Lectures—Physiology or Medicine 1942–1962. (Amsterdam: Elsevier Publishing Company), pp. 186-206. Abstract Article
Correa, P.A., Gruninger, T., and García, L.R. (2015). DOP-2 D2-like receptor regulates UNC-7 innexins to attenuate recurrent sensory motor neurons during C. elegans copulation. J. Neurosci. 35, 9990-10004. Abstract Article
Correa, P., LeBoeuf, B., and García, L.R. (2012). C. elegans dopaminergic D2-like receptors delimit recurrent cholinergic-mediated motor programs during a goal-oriented behavior. PLoS Genet. 8, e1003015. Abstract Article
Cuppen, E., van der Linden, A.M., Jansen, G., and Plasterk, R.H. (2003). Proteins interacting with Caenorhabditis elegans Gα subunits. Comp. Funct. Genomics 4, 479-491. Abstract Article
de Bono, M., and Bargmann, C.I. (1998). Natural variation in a neuropeptide Y receptor homolog modifies social behavior and food response in C. elegans. Cell 94, 679-689. Abstract Article
Dempsey, C.M., Mackenzie, S.M., Gargus, A., Blanco, G., Sze, J.Y. (2005). Serotonin (5HT), fluoxetine, imipramine and dopamine target distinct 5HT receptor signaling to modulate Caenorhabditis elegans egg-laying behavior. Genetics 169, 1425-1436. Abstract Article
Dernovici, S., Starc, T., Dent, J.A., and Ribeiro, P. (2007). The serotonin receptor SER-1 (5HT2ce) contributes to the regulation of locomotion in Caenorhabditis elegans. Dev. Neurobiol. 67, 189-204. Abstract Article
Desai, C., and Horvitz, H.R. (1989). Caenorhabditis elegans mutants defective in the functioning of the motor neurons responsible for egg laying. Genetics 121, 703-721. Abstract Article
Dietzel, C., and Kurjan, J. (1987). Pheromonal regulation and sequence of the Saccharomyces cerevisiae SST2 gene: a model for desensitization to pheromone. Mol. Cell Biol. 7, 4169-4177. Abstract Article
Dillon, J., Franks, C.J., Murray, C., Edwards, R.J., Calahorro, F., Ishihara, T., Katsura, I., Holden-Dye, L., and O'Connor, V. (2015). Metabotropic glutamate receptors: modulators of context-dependent feeding behavior in C. elegans. J. Biol. Chem. 290, 15052-15065. Abstract Article
Dittman, J.S., and Kaplan, J.M. (2008). Behavioral impact of neurotransmitter-activated G-protein-coupled receptors: muscarinic and GABAB receptors regulate Caenorhabditis elegans locomotion. J. Neurosci. 28, 7104-7112. Abstract Article
Dohlman, H.G., and Thorner, J.W. (2001). Regulation of G protein-initiated signal transduction in yeast: paradigms and principles. Annu. Rev. Biochem. 70, 703-754. Abstract Article
Dohlman, H.G., Song, J., Ma, D., Courchesne, W.E., Thorner, J. (1996). Sst2, a negative regulator of pheromone signaling in the yeast Saccharomyces cerevisiae: expression, localization, and genetic interaction and physical association with Gpa1 (the G-protein α subunit). Mol. Cell Biol. 16, 5194-5209. Abstract Article
Donaldson, Z.R., Nautiyal, K.M., Ahmari, S.E., and Hen. R. (2013). Genetic approaches for understanding the role of serotonin receptors in mood and behavior. Curr. Opin. Neurobiol. 23, 399-406. Abstract Article
Dong, M.Q., Chase, D., Patikoglou, G.A., and Koelle, M.R. (2000). Multiple RGS proteins alter neural G protein signaling to allow C. elegans to rapidly change behavior when fed. Genes Dev. 14, 2003-2014. Abstract Article
Donnelly, J.L., Clark, C.M., Leifer, A.M., Pirri, J.K., Haburcak, M., Francis, M.M., Samuel, A.D., and Alkema, M.J. (2013). Monoaminergic orchestration of motor programs in a complex C. elegans behavior. PLoS Biol. 11, e1001529. Abstract Article
Donohoe, D.R., Jarvis, R.A., Weeks, K., Aamodt, E.J., and Dwyer, D.S. (2009). Behavioral adaptation in C. elegans produced by antipsychotic drugs requires serotonin and is associated with calcium signaling and calcineurin inhibition. Neurosci. Res. 64, 280-289. Abstract Article
Emtage, L., Aziz-Zaman, S., Padovan-Merhar, O., Horvitz, H.R., Fang-Yen, C., and Ringstad, N. (2012). IRK-1 potassium channels mediate peptidergic inhibition of Caenorhabditis elegans serotonin neurons via a Go signaling pathway. J. Neurosci. 32, 16285-16295. Abstract Article
Ezak, M.J., and Ferkey, D.M. (2010). The C. elegans D2-like dopamine receptor DOP-3 decreases behavioral sensitivity to the olfactory stimulus 1-octanol. PLoS One 5, e9487. Abstract Article
Ezcurra, M., Tanizawa, Y., Swoboda, P., and Schafer, W.R. (2011). Food sensitizes C. elegans avoidance behaviours through acute dopamine signalling. EMBO J. 30, 1110-1122. Abstract Article
Fantegrossi, W.E., Murnane, K.S., and Reissig, C.J. (2008). The behavioral pharmacology of hallucinogens. Biochem. Pharmacol. 75, 17-33. Abstract Article
Ferkey, D.M., Hyde, R., Haspel, G., Dionne, H.M., Hess, H.A., Suzuki, H., Schafer, W.R., Koelle, M.R., and Hart, A.C. (2007). C. elegans G protein regulator RGS-3 controls sensitivity to sensory stimuli. Neuron 53, 39-52. Abstract Article
Fischer, E.H. (1992). Protein Phosphorylation and Cellular Regulation, II. In Nobel Lectures, Physiology or Medicine 1991-1995. N. Ringertz, ed. (Singapore: World Scientific Publishing Co.), pp. 95-113. Article
Frooninckx, L., Van Rompay, L., Temmerman, L., Van Sinay, E., Beets, I., Janssen, T.,Husson, S.J., and Schoofs, L. (2012). Neuropeptide GPCRs in C. elegans. Front. Endocrinol. 3, 167. Abstract Article
Fukuto, H.S., Ferkey, D.M., Apicella, A.J., Lans, H., Sharmeen, T., Chen, W., Lefkowitz, R.J., Jansen, G., Schafer, W.R., and Hart, A.C. (2004). G protein-coupled receptor kinase function is essential for chemosensation in C. elegans. Neuron 42, 581-593. Abstract Article
Fung, B.K., Hurley, J.B., and Stryer, L. (1981). Flow of information in the light-triggered cyclic nucleotide cascade of vision. Proc. Natl. Acad. Sci. U. S. A. 78, 152-156. Abstract Article
Gabay, M., Pinter, M.E., Wright, F.A., Chan, P., Murphy, A.J., Valenzuela, D.M., Yancopoulos, G.D., and Tall, G.G. (2011). Ric-8 proteins are molecular chaperones that direct nascent G protein α subunit membrane association. Sci. Signal. 4, ra79. Abstract Article
Ghosh-Roy, A., Wu, Z., Goncharov, A., Jin, Y., and Chisholm, A.D. (2010). Calcium and cyclic AMP promote axonal regeneration in Caenorhabditis elegans and require DLK-1 kinase. J. Neurosci. 30, 3175-3183. Abstract Article
Gilman, A.G. (1994). G Proteins and Regulation of Adenylyl Cyclase. In Nobel Lectures – Physiology or Medicine 1991–1995. N. Ringertz, ed. (Singapore: World Scientific Publishing Co.), pp. 182-212. Article
Godinho, R.O., Duarte, T., and Pacini, E.S. (2015). New perspectives in signaling mediated by receptors coupled to stimulatory G protein: the emerging significance of cAMP efflux and extracellular cAMP-adenosine pathway. Front. Pharmacol. 6, 58. Abstract Article
Gomez-Amaro, R.L., Valentine, E.R., Carretero, M., LeBoeuf, S.E., Rangaraju, S., Broaddus, C.D., Solis, G.M., Williamson, J.R., and Petrascheck, M. (2015). Measuring food intake and nutrient absorption in Caenorhabditis elegans. Genetics 200, 443-454. Abstract Article
González-Maeso, J. (2011). GPCR oligomers in pharmacology and signaling. Mol. Brain 4, 20. Abstract Article
Gotta, M., and Ahringer J. (2001). Distinct roles for Gα and Gβγ in regulating spindle position and orientation in Caenorhabditis elegans embryos. Nat. Cell Biol. 3, 297-300. Abstract Article
Gotta, M., Dong, Y., Peterson, Y.K., Lanier, S.M., and Ahringer, J. (2003). Asymmetrically distributed C. elegans homologs of AGS3/PINS control spindle position in the early embryo. Curr. Biol. 13, 1029-1037. Abstract Article
Govindan, J.A., Cheng, H., Harris, J.E., and Greenstein, D. (2006). Gαo/i and Gαs signaling function in parallel with the MSP/Eph receptor to control meiotic diapause in C. elegans. Curr. Biol. 16, 1257-1268. Abstract Article
Guo, M., Wu, T.H., Song, Y.X., Ge, M.H., Su, C.M., Niu, W.P., Li, L.L., Xu, Z.J., Ge, C.L., Al-Mhanawi., M.T., Wu, S.P., and Wu, Z.X. (2015). Reciprocal inhibition between sensory ASH and ASI neurons modulates nociception and avoidance in Caenorhabditis elegans. Nat. Commun. 6, 5655. Abstract Article
Gürel, G., Gustafson, M.A., Pepper, J.S., Horvitz, H.R., and Koelle, M.R. (2012). Receptors and other signaling proteins required for serotonin control of locomotion in Caenorhabditis elegans. Genetics 192, 1359-1371. Abstract Article
Hajdu-Cronin, Y.M., Chen, W.J., Patikoglou, G., Koelle, M.R., and Sternberg, P.W. (1999). Antagonism between Goα and Gqα in Caenorhabditis elegans: the RGS protein EAT-16 is necessary for Goα signaling and regulates Goα activity. Genes Dev. 3, 1780-1793. Abstract Article
Hall, R.A. (2004). β-adrenergic receptors and their interacting proteins. Semin. Cell Dev. Biol. 15, 281-288. Abstract Article
Hamdan, F.F., Ungrin, M.D., Abramovitz, M., and Ribeiro, P. (1999). Characterization of a novel serotonin receptor from Caenorhabditis elegans: cloning and expression of two splice variants. J. Neurochem. 72, 1372-1383. Abstract Article
Hapiak, V.M., Hobson, R.J., Hughes, L., Smith, K., Harris, G., Condon, C., Komuniecki, P., and Komuniecki, R.W. (2009). Dual excitatory and inhibitory serotonergic inputs modulate egg laying in Caenorhabditis elegans. Genetics 181, 153-163. Abstract Article
Hapiak, V., Summers, P., Ortega, A., Law, W.J., Stein, A., and Komuniecki, R. (2013). Neuropeptides amplify and focus the monoaminergic inhibition of nociception in Caenorhabditis elegans. J. Neurosci. 33, 14107-14116. Abstract Article
Harris, G.P., Hapiak, V.M., Wragg, R.T., Miller, S.B., Hughes, L.J., Hobson, R.J., Steven, R, Bamber, B., and Komuniecki, R.W. (2009). Three distinct amine receptors operating at different levels within the locomotory circuit are each essential for the serotonergic modulation of chemosensation in Caenorhabditis elegans. J. Neurosci. 29, 1446-1456. Abstract Article
Harris, G., Korchnak, A., Summers, P., Hapiak, V., Law, W.J., Stein, A.M., Komuniecki, P., and Komuniecki, R. (2011). Dissecting the serotonergic food signal stimulating sensory-mediated aversive behavior in C. elegans. PLoS One. 6, e21897. Abstract Article
Harris, G., Mills, H., Wragg, R., Hapiak, V., Castelletto, M., Korchnak, A., and Komuniecki, R.W. (2010). The monoaminergic modulation of sensory-mediated aversive responses in Caenorhabditis elegans requires glutamatergic/peptidergic cotransmission. J. Neurosci. 30, 7889-7899. Abstract Article
Hendricks, M., Ha, H., Maffey, N., and Zhang, Y. (2012). Compartmentalized calcium dynamics in a C. elegans interneuron encode head movement. Nature 487, 99-103. Abstract Article
Hepler, J.R., and Gilman, .AG. (1992). G proteins. Trends Biochem. Sci. 17, 383-387. Abstract Article
Herlitze, S., Garcia, D.E., Mackie, K., Hille, B., Scheuer, T., and Catterall, W.A. (1996). Modulation of Ca2+ channels by G-protein βγ subunits. Nature 380, 258-262. Abstract Article
Hess, H.A., Röper, J.C., Grill, S.W., and Koelle, M.R. (2004). RGS-7 completes a receptor-independent heterotrimeric G protein cycle to asymmetrically regulate mitotic spindle positioning in C. elegans. Cell 119, 209-218. Abstract Article
Hiley, E., McMullan, R., and Nurrish, S.J. (2006). The Gα12-RGS RhoGEF-RhoA signaling pathway regulates neurotransmitter release in C. elegans. EMBO J. 25, 5884-5895. Abstract Article
Hobert O. The neuronal genome of Caenorhabditis elegans (August 13, 2013), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.161.1, http://www.wormbook.org. Abstract Article
Hobson, R.J., Geng, J., Gray, A.D., and Komuniecki, R.W. (2003). SER-7b, a constitutively active Gαs coupled 5-HT7-like receptor expressed in the Caenorhabditis elegans M4 pharyngeal motorneuron. J. Neurochem. 87, 22-29. Abstract Article
Hobson, R.J., Hapiak, V.M., Xiao, H., Buehrer, K.L., Komuniecki, P.R., and Komuniecki, R.W. (2006). SER-7, a Caenorhabditis elegans 5-HT7-like receptor, is essential for the 5-HT stimulation of pharyngeal pumping and egg laying. Genetics 172, 159-169. Abstract Article
Hofler, C., and Koelle, M.R. (2011). AGS-3 alters Caenorhabditis elegans behavior after food deprivation via RIC-8 activation of the neural G protein Gαo. J. Neurosci. 31, 11553-11562. Abstract Article
Hofler, C., and Koelle, M.R. (2012). The G protein regulator AGS-3 allows C. elegans to alter behaviors in response to food deprivation. Worm 1, 56-60. Abstract Article
Hoyer, D., and Bartfai, T. (2012). Neuropeptides and neuropeptide receptors: drug targets, and peptide and non-peptide ligands: a tribute to Prof. Dieter Seebach. Chem. Biodivers. 9, 2367-2387. Abstract Article
Hu, Z., Pym, E.C., Babu, K., Vashlishan Murray, A.B., and Kaplan, J.M. (2011). A neuropeptide-mediated stretch response links muscle contraction to changes in neurotransmitter release. Neuron 71, 92-102. Abstract Article
Hu, Z., Vashlishan-Murray, A.B., and Kaplan, J.M. (2015). NLP-12 engages different UNC-13 proteins to potentiate tonic and evoked release. J. Neurosci. 35, 1038-1042. Abstract Article
Hwang, J.M., Chang, D.J., Kim, U.S., Lee, Y.S., Park, Y.S., Kaang, B.K., and Cho, N.J. (1999). Cloning and functional characterization of a Caenorhabditis elegans muscarinic acetylcholine receptor. Receptors Channels 6, 415-424. Abstract Article
Jafari, G., Xie, Y., Kullyev, A., Liang, B., and Sze, J.Y. (2011). Regulation of extrasynaptic 5-HT by serotonin reuptake transporter function in 5-HT-absorbing neurons underscores adaptation behavior in Caenorhabditis elegans. J. Neurosci. 15, 8948-8957. Abstract Article
James, D.J., and Martin, T.F. (2013). CAPS and Munc13: CATCHRs that SNARE vesicles. Front. Endocrinol. 4, 187. Abstract Article
Jansen, G., Thijssen, K.L., Werner, P., van der Horst, M., Hazendonk, E., and Plasterk R.H. (1999). The complete family of genes encoding G proteins of Caenorhabditis elegans. Nat. Genet. 21, 414-419. Abstract Article
Jansen, G., Weinkove, D., and Plasterk, R.H. (2002). The G-protein γ subunit gpc-1 of the nematode C. elegans is involved in taste adaptation. EMBO J. 21, 986-994. Abstract Article
Janssen, T., Husson, S.J., Lindemans, M., Mertens, I., Rademakers, S., Ver Donck, K., Geysen, J., Jansen, G., and Schoofs, L. (2008). Functional characterization of three G protein-coupled receptors for pigment dispersing factors in Caenorhabditis elegans. J. Biol. Chem. 283, 15241-15239. Abstract Article
Joffe, M.E., Grueter, C.A., and Grueter, B.A. (2014). Biological substrates of addiction. Wiley Interdiscip. Rev. Cogn. Sci. 5, 151-171. Abstract Article
Jorgensen, E.M. GABA (August 31, 2005), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.14.1, http://www.wormbook.org. Abstract Article
Jorgenson, L.A., Newsome, W.T., Anderson, D.J., Bargmann. C.I., Brown, E.N., Deisseroth, K., Donoghue, J.P., Hudson, K.L., Ling, G.S., MacLeish, P.R., et al. (2015). The BRAIN Initiative: developing technology to catalyse neuroscience discovery. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370, pii: 20140164. Abstract Article
Jose, A.M., and Koelle, M.R. (2005). Domains, amino acid residues, and new isoforms of Caenorhabditis elegans diacylglycerol kinase 1 (DGK-1) important for terminating diacylglycerol signaling in vivo. J. Biol. Chem. 280, 2730-2736. Abstract Article
Jose, A.M., Bany, I.A., Chase, D.L., and Koelle, M.R. (2007). A specific subset of transient receptor potential vanilloid-type channel subunits in Caenorhabditis elegans endocrine cells function as mixed heteromers to promote neurotransmitter release. Genetics 175, 93-105. Abstract Article
Kadamur, G., and Ross, E.M. (2013). Mammalian phospholipase C. Annu. rev. Physiol. 75, 127-154. Abstract Article
Kandel E.R. (2004). The molecular biology of memory storage: a dialog between genes and synapses. Biosci. Rep. 24, 475-522. Abstract Article
Kandel, E.R. (2012). The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol. Brain. 5, 14. Abstract Article
Kang, C., and Avery, L. (2009). Systemic regulation of starvation response in Caenorhabditis elegans. Genes Dev. 23, 12-17. Abstract Article
Kanner, B.I., and Zomot, E. (2008). Sodium-coupled neurotransmitter transporters. Chem. Rev. 108, 1654-1668. Abstract Article
Katritch, V., Cherezov, V., and Stevens, R.C. (2013). Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 53, 531-556. Abstract Article
Kauffman, A.L., Ashraf, J.M., Corces-Zimmerman, M.R., Landis, J.N., and Murphy, C.T. (2010). Insulin signaling and dietary restriction differentially influence the decline of learning and memory with age. PLoS Biol. 8, e1000372. Abstract Article
Kaupmann, K., Malitschek, B., Schuler, V., Heid, J., Froestl, W., Beck, P., Mosbacher, J., Bischoff, S., Kulik, A., Shigemoto, R., et al. (1998). GABAB-receptor subtypes assemble into functional heteromeric complexes. Nature 396, 683-687. Abstract Article
Keating, C.D., Kriek, N., Daniels, M., Ashcroft, N.R., Hopper, N.A., Siney, E.J., Holden- Dye, L., and Burke, J.F. (2003). Whole-genome analysis of 60 G protein-coupled receptors in Caenorhabditis elegans by gene knockout with RNAi. Curr. Biol. 13, 1715-1720. Abstract Article
Khan, S.M., Sleno, R., Gora, S., Zylbergold, P., Laverdure, J.P., Labbé, J.C., Miller, G.J., and Hébert, T.E. (2013). The expanding roles of Gβγ subunits in G protein-coupled receptor signaling and drug action. Pharmacol. Rev. 65, 545-577. Abstract Article
Kida, S., Josselyn, S.A., Peña de Ortiz, S., Kogan, J.H., Chevere, I., Masushige, S., and Silva, A.J. (2002). CREB required for the stability of new and reactivated fear memories. Nat. Neurosci. 5, 348-355. Abstract Article
Kimura, K.D., Fujita, K., and Katsura, I. (2010). Enhancement of odor avoidance regulated by dopamine signaling in Caenorhabditis elegans. J. Neurosci. 30, 16365-16375. Abstract Article
Kindt, K.S., Quast, K.B., Giles, A.C., De, S., Hendrey, D., Nicastro, I., Rankin, C.H., and Schafer, W.R. (2007). Dopamine mediates context-dependent modulation of sensory plasticity in C. elegans. Neuron 55, 662-676. Abstract Article
Kobilka. B. (2013). The Structural Basis of G-Protein-Coupled Receptor Signaling (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 52, 6380-6288. Abstract Article
Koelle, M.R., and Horvitz, H.R. (1996). EGL-10 regulates G protein signaling in the C. elegans nervous system and shares a conserved domain with many mammalian proteins. Cell 84, 115-125. Abstract Article
Kohout, T.A., and Lefkowitz, R.J. (2003). Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol. Pharmacol. 63, 9-18. Abstract Article
Komuniecki, R.W., Hobson, R.J., Rex, E.B., Hapiak, V.M., and Komuniecki, P.R. (2004). Biogenic amine receptors in parasitic nematodes: what can be learned from Caenorhabditis elegans? Mol. Biochem. Parasitol. 137, 1-11. Abstract Article
Korswagen, H.C., Park, J.H., Ohshima, Y., and Plasterk, R.H. (1997). An activating mutation in a Caenorhabditis elegans Gs protein induces neural degeneration. Genes Dev. 11, 1493-1503. Abstract Article
Krebs, E.G. (1992). Protein Phosphorylation and Cellular Regulation, I. In Nobel Lectures, Physiology or Medicine 1991-1995. N. Ringertz, ed. (Singapore: World Scientific Publishing Co.), pp. 72-89. Abstract Article
Kruse, A.C., Kobilka, B.K., Gautam, D., Sexton, P.M., Christopoulos, A., and Wess. J. (2014). Muscarinic acetylcholine receptors: novel opportunities for drug development. Nat. Rev. Drug Discov. 13, 549-560. Abstract Article
Kubiak, T.M., Larsen, M.J., Nulf, S.C., Zantello, M.R., Burton, K.J., Bowman, J.W., Modric, T., and Lowery, D.E. (2003). Differential activation of “social” and “solitary” variants of the Caenorhabditis elegans G protein-coupled receptor NPR-1 by its cognate ligand AF9. J. Biol. Chem. 278, 33724-33729. Abstract Article
Kullyev, A., Dempsey, C.M, Miller, S., Kuan, C.J., Hapiak, V.M., Komuniecki, R.W., Griffin,, C.T., and Sze, J.Y. (2010). A genetic survey of fluoxetine action on synaptic transmission in Caenorhabditis elegans. Genetics 186, 929-941. Abstract Article
Kuromi, H., and Kidokoro, Y. (2000). Tetanic stimulation recruits vesicles from reserve pool via a cAMP-mediated process in Drosophila synapses. Neuron 27, 133-143. Abstract Article
Lackner, M.R., Nurrish, S.J., and Kaplan, J.M. (1999). Facilitation of synaptic transmission by EGL-30 Gqα and EGL-8 PLCβ: DAG binding to UNC-13 is required to stimulate acetylcholine release. Neuron 24, 335-346. Abstract Article
Lambright, D.G., Noel, J.P., Hamm, H.E., and Sigler, P.B. (1994). Structural determinants for activation of the α-subunit of a heterotrimeric G protein. Nature 369, 621-628. Abstract Article
Lee, Y.S., Park, Y.S., Nam, S., Suh, S.J., Lee, J., Kaang, B.K., and Cho, N.J. (2000). Characterization of GAR-2, a novel G protein-linked acetylcholine receptor from Caenorhabditis elegans. J. Neurochem. 75, 1800-1809. Abstract Article
Lefkowitz, R.J. (2013). A Brief History of G-Protein Coupled Receptors (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 52, 6366-6378. Abstract Article
Leiser, S.F., Miller, H., Rossner, R., Fletcher, M., Leonard, A., Primitivo, M., Rintala, N., Ramos, F.J., Miller, D.L., and Kaeberlein, M. (2015). Cell nonautonomous activation of flavin-containing monooxygenase promotes longevity and health span. Science 350, 1375-1378. Abstract Article
Lemoine, D., Jiang, R., Taly, A., Chataigneau, T., Specht, A., and Grutter, T. (2012). Ligand-gated ion channels: new insights into neurological disorders and ligand recognition. Chem. Rev. 112, 6285-6318. Abstract Article
Li, C. and Kim, K. Neuropeptides (April 27, 2008) WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.142.1, http://www.wormbook.org. Abstract Article
Li, C., and Kim, K. (2010). Neuropeptide gene families in Caenorhabditis elegans. Adv. Exp. Med. Biol. 692, 98-137. Abstract Article
Li, G., and Zhang, X.C. (2004). GTP hydrolysis mechanism of Ras-like GTPases. J. Mol. Biol. 340, 921-932. Abstract Article
Li, Y., Zhao, Y., Huang, X., Lin, X., Guo, Y., Wang, D., Li, C., and Wang, D. (2013). Serotonin control of thermotaxis memory behavior in nematode Caenorhabditis elegans. PLoS One 8, e77779. Abstract Article
Liu, Y., LeBoeuf, B., and Garcia, L.R. (2007). Gαq-coupled muscarinic acetylcholine receptors enhance nicotinic acetylcholine receptor signaling in Caenorhabditis elegans mating behavior. J. Neurosci. 27, 1411-1421. Abstract Article
Love, T.M. (2014). Oxytocin, motivation and the role of dopamine. Pharmacol. Biochem. Behav. 119, 49-60. Abstract Article
Lutz, S., Shankaranarayanan, A., Coco, C., Ridilla, M., Nance, M.R., Vettel, C., Baltus, D., Evelyn, C.R., Neubig, R.R., Wieland, T., and Tesmer, J.J. (2007). Structure of Gαq-p63RhoGEF-RhoA complex reveals a pathway for the activation of RhoA by GPCRs. Science 318, 1923-1927. Abstract Article
Marder, E. (2012). Neuromodulation of neuronal circuits: back to the future. Neuron 76, 1-11. Abstract Article
Matsuki, M., Kunitomo, H., and Iino, Y. (2006). Goα regulates olfactory adaptation by antagonizing Gqα-DAG signaling in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 103, 1112-1117. Abstract Article
McDonald, P.W., Hardie, S.L., Jessen, T.N., Carvelli, L., Matthies, D.S., and Blakely, R.D. (2007). Vigorous motor activity in Caenorhabditis elegans requires efficient clearance of dopamine mediated by synaptic localization of the dopamine transporter DAT-1. J. Neurosci. 27, 14216-14227. Abstract Article
McMullan, R., Hiley, E., Morrison, P., Nurrish, S.J. (2006). Rho is a presynaptic activator of neurotransmitter release at pre-existing synapses in C. elegans. Genes Dev. 20, 65-76. Abstract Article
Mendel, J.E., Korswagen, H.C., Liu, K.S., Hajdu-Cronin, Y.M., Simon, M.I., Plasterk, R.H., and Sternberg, P.W. (1995). Participation of the protein Go in multiple aspects of behavior in C. elegans. Science 267, 1652-1655. Abstract Article
Merighi, A., Salio, C., Ferrini, F., and Lossi, L. (2011). Neuromodulatory function of neuropeptides in the normal CNS. J. Chem. Neuroanat. 42, 276-287. Abstract Article
Mersha, M., Formisano, R., McDonald, R., Pandey, P., Tavernarakis, N., and Harbinder, S. (2013). GPA-14, a Gαi subunit mediates dopaminergic behavioral plasticity in C. elegans. Behav. Brain Funct. 9, 16. Abstract Article
Mertens, I., Vandingenen, A., Meeusen, T., De Loof, A., and Schoofs, L. (2004). Postgenomic characterization of G-protein-coupled receptors. Pharmacogenomics 5, 657-672. Abstract Article
Miller, K.G., and Rand, J.B. (2000). A role for RIC-8 (Synembryn) and GOA-1 (Goα) in regulating a subset of centrosome movements during early embryogenesis in Caenorhabditis elegans. Genetics 156, 1649-1660. Abstract Article
Miller, K.G., Alfonso, A., Nguyen, M., Crowell, J.A., Johnson, C.D., and Rand, J.B. (1996). A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc. Natl. Acad. Sci. U. S. A. 93, 12593-12598. Abstract Article
Miller, K.G., Emerson, M.D., and Rand, J.B. (1999). Goα and diacylglycerol kinase negatively regulate the Gqα pathway in C. elegans. Neuron. 24, 323-233. Abstract Article
Miller, K.G, Emerson, M.D., McManus, J.R., and Rand, J.B. (2000). RIC-8 (Synembryn): a novel conserved protein that is required for Gqα signaling in the C. elegans nervous system. Neuron 27, 289-299. Abstract Article
Mills, H., Wragg, R., Hapiak, V., Castelletto, M., Zahratka, J., Harris, G., Summers, P., Korchnak, A., Law, W., Bamber, B., and Komuniecki, R. (2012). Monoamines and neuropeptides interact to inhibit aversive behaviour in Caenorhabditis elegans. EMBO J. 31, 667-678. Abstract Article
Min, D.S., Cho, N.J., Yoon, S.H., Lee, Y.H., Hahn, S.J., Lee, K.H., Kim, M.S., and Jo, Y.H. (2000). Phospholipase C, protein kinase C, Ca2+/calmodulin-dependent protein kinase II, and tyrosine phosphorylation are involved in carbachol-induced phospholipase D activation in Chinese hamster ovary cells expressing muscarinic acetylcholine receptor of Caenorhabditis elegans. J. Neurochem. 75, 274-281. Abstract Article
Moorman, C., and Plasterk, R.H. (2002). Functional characterization of the adenylyl cyclase gene sgs-1 by analysis of a mutational spectrum in Caenorhabditis elegans. Genetics. 161, 133-142. Abstract Article
Moreira, I.S. (2014). Structural features of the G-protein/GPCR interactions. Biochim. Biophys. Acta 1840, 16-33. Abstract Article
Murakami, H., and Murakami S. (2007). Serotonin receptors antagonistically modulate Caenorhabditis elegans longevity. Aging Cell 6, 483-438. Abstract Article
Näsman, J., Kukkonen, J.P., Holmqvist, T., and Akerman, K.E. (2002). Different roles for Gi and Go proteins in modulation of adenylyl cyclase type-2 activity. J. Neurochem. 83, 1252-1261. Abstract Article
Nelson, J.C., and Colón-Ramos, D.A. (2012). Serotonergic neurosecretory synapse targeting is controlled by netrin-releasing guidepost neurons in Caenorhabditis elegans. J. Neurosci. 33, 1366-1376. Abstract Article
Noel, J.P., Hamm, H.E., and Sigler, P.B. (1993). The 2.2 Å crystal structure of transducin-α complexed with GTPγS. Nature 366, 654-663. Abstract Article
Nurrish, S., Ségalat, L., and Kaplan, J.M. (1999). Serotonin inhibition of synaptic transmission: Gαo decreases the abundance of UNC-13 at release sites. Neuron 24, 231-242. Abstract Article
Okada, T., Kajimoto, T., Jahangeer, S., and Nakamura, S. (2009). Sphingosine kinase/sphingosine 1-phosphate signalling in central nervous system. Cell Signal. 21, 7-13. Abstract Article
Olde, B., and McCombie W.R. (1997). Molecular cloning and functional expression of a serotonin receptor from Caenorhabditis elegans. J. Mol. Neurosci. 8, 53-62. Abstract Article
Omura, D.T., Clark, D.A., Samuel, A.D., and Horvitz, H.R. (2012). Dopamine signaling is essential for precise rates of locomotion by C. elegans. PLoS One 7, e38649. Abstract Article
Palmitessa, A., Hess, H.A., Bany, I.A., Kim, Y.M., Koelle, M.R., and Benovic, J.L. (2005). Caenorhabditus elegans arrestin regulates neural G protein signaling and olfactory adaptation and recovery. J. Biol. Chem. 280, 24649-24662. Abstract Article
Pandey, P., and Harbinder, S. (2012). The Caenorhabditis elegans D2-like dopamine receptor DOP-2 physically interacts with GPA-14, a Gαi subunit. J. Mol. Signal. 7, 3. Abstract Article
Park, D., O’Doherty, I., Somvanshi, R.K., Bethke, A., Schroeder, F.C., Kumar, U., and Riddle, D.L. (2012). Interaction of structure-specific and promiscuous G-protein-coupled receptors mediates small-molecule signaling in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 109, 9917-9922. Abstract Article
Park, J.H., Ohshima, S., Tani, T., Ohshima, Y. (1997). Structure and expression of the gsa-1 gene encoding a G protein αs subunit in C. elegans. Gene 194, 183-190. Abstract Article
Park, Y.S., Cho, T.J., and Cho, N.J. (2006). Stimulation of cyclic AMP production by the Caenorhabditis elegans muscarinic acetylcholine receptor GAR-3 in Chinese hamster ovary cells. Arch. Biochem. Biophys. 450, 203-207. Abstract Article
Park, Y.S., Kim, S., Shin, Y., Choi, B., and Cho, N.J. (2003). Alternative splicing of the muscarinic acetylcholine receptor GAR-3 in Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 308, 961-965. Abstract Article
Park, Y.S., Lee, Y.S., Cho, N.J., and Kaang, B.K. (2000). Alternative splicing of gar-1, a Caenorhabditis elegans G-protein-linked acetylcholine receptor gene. Biochem. Biophys. Res. Commun. 268, 354-358. Abstract Article
Patikoglou, G.A., and Koelle, M.R. (2002). An N-terminal region of Caenorhabditis elegans RGS proteins EGL-10 and EAT-16 directs inhibition of Gαo versus Gαq signaling. J. Biol. Chem. 277, 47004-47013. Abstract Article
Perazzona, B., Isabel, G., Preat, T., and Davis, R.L. (2004). The role of cAMP response element-binding protein in Drosophila long-term memory. J. Neurosci. 24, 8823-8828. Abstract Article
Pereira, L., Kratsios, P., Serrano-Saiz, E., Sheftel, H., Mayo, A.E., Hall, D.H., White, J.G., LeBoeuf. B., Garcia, L.R., Alon, U., and Hobert, O. (2015). A cellular and regulatory map of the cholinergic nervous system of C. elegans. eLife 4, pii: e12432. Abstract Article
Pereira, S., and van der Kooy, D. (2012). Two forms of learning following training to a single odorant in Caenorhabditis elegans AWC neurons. J. Neurosci. 32, 9035-9044. Abstract Article
Perez-Mansilla, B, and Nurrish, S. (2009). A network of G-protein signaling pathways control neuronal activity. Adv. Genet. 65, 145-192. Abstract Article
Peterson, Y.K., Bernard, M.L., Ma, H., Hazard, S. III, Graber, S.G., and Lanier, S.M. (2000). Stabilization of the GDP-bound conformation of Giα by a peptide derived from the G-protein regulatory motif of AGS3. J. Biol. Chem. 275, 33193-33196. Abstract Article
Petrascheck, M., Ye, X., and Buck, L.B. (2007). An antidepressant that extends lifespan in adult Caenorhabditis elegans. Nature 450, 553-556. Abstract Article
Peymen, K., Watteyne, J., Frooninckx, L., Schoofs, L., and Beets, I. (2014). The FMRFamide-like peptide family in nematodes. Front. Endocrinol. 5, 90. Abstract Article
Pirri, J.K., McPherson, A.D., Donnelly, J.L., Francis, M.M., and Alkema, M.J. (2009). A tyramine-gated chloride channel coordinates distinct motor programs of a Caenorhabditis elegans escape response. 62, 526-538. Abstract Article
Pocock, R., and Hobert, O. (2010). Hypoxia activates a latent circuit for processing gustatory information in C. elegans. Nat. Neurosci. 13, 610-614. Abstract Article
Poole, R.J., Bashllari, E., Cochella, L., Flowers, E.B., and Hobert, O. (2011). A genome-wide RNAi screen for factors involved in neuronal specification in Caenorhabditis elegans. PLoS Genet. 7, e1002109. Abstract Article
Porter, M.Y., and Koelle, M.R. (2009). Insights into RGS protein function from studies in Caenorhabditis elegans. Prog. Mol. Biol. Transl. Sci. 86, 15-47. Abstract Article
Pytliak, M., Vargová, V., Mechírová, V., and Felšöci, M. (2011). Serotonin receptors—from molecular biology to clinical applications. Physiol. Res. 60, 15-25. Abstract Article
Rand, J.B. Acetylcholine (January 30, 2007), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.131.1, http://www.wormbook.org. Abstract Article
Rasmussen, S.G., DeVree, B.T., Zou, Y., Kruse, A.C., Chung, K.Y., Kobilka, T.S., Thian, F.S., Chae, P.S., Pardon, E., Calinski, D., et al. (2011). Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549-555. Abstract Article
Reuveny, E., Slesinger, P.A., Inglese, J., Morales, J.M., Iñiguez-Lluhi, J.A., Lefkowitz, R.J., Bourne, H.R., Jan, Y.N., and Jan, L.Y. (1994). Activation of the cloned muscarinic potassium channel by G protein βγ subunits. Nature 370, 143-146. Abstract Article
Rex, E., and Komuniecki, R.W. (2002). Characterization of a tyramine receptor from Caenorhabditis elegans. J. Neurochem. 82, 1352-1359. Abstract Article
Rex, E., Hapiak, V., Hobson, R., Smith, K., Xiao, H., and Komuniecki, R. (2005). TYRA-2 (F01E11.5): a Caenorhabditis elegans tyramine receptor expressed in the MC and NSM pharyngeal neurons. J. Neurochem. 94, 181-189 Abstract Article
Rex, E., Molitor, S.C., Hapiak, V., Xiao, H., Henderson, M., and Komuniecki, R. (2004). Tyramine receptor (SER-2) isoforms are involved in the regulation of pharyngeal pumping and foraging behavior in Caenorhabditis elegans. J. Neurochem. 91, 1104-1115. Abstract Article
Reynolds, N.K., Schade, M.A., and Miller, K.G. (2005). Convergent, RIC-8-dependent Gα signaling pathways in the Caenorhabditis elegans synaptic signaling network. Genetics 169, 651-670. Abstract Article
Rhee, J.S., Betz, A., Pyott, S., Reim, K., Varoqueaux, F., Augustin, I., Hesse, D., Robatzek, M., Niacaris, T., Steger, K., Avery, L., and Thomas, J.H. (2001). β phorbol ester- and diacylglycerol-induced augmentation of transmitter release is mediated by Munc13s and not by PKCs. Cell 108, 121-133. Abstract Article
Ringstad, N., and Horvitz, H.R. (2008). FMRFamide neuropeptides and acetylcholine synergistically inhibit egg-laying by C. elegans. Nat. Neurosci. 11, 1168-1176. Abstract Article
Robatzek, M., Niacaris, T., Steger, K., Avery, L., and Thomas, J.H. (2001). eat-11 encodes GPB-2, a Gβ5 ortholog that interacts with Goα and Gqα to regulate C. elegans behavior. Curr. Biol. 11, 288-293. Abstract Article
Rodbell, M. (1994). Signal Transduction: Evolution of an Idea. In Nobel Lectures – Physiology or Medicine 1991–1995. N. Ringertz, ed. (Singapore: World Scientific Publishing Co.), pp. 220-237. Abstract Article
Rogers, C., Reale, V., Kim, K., Chatwin, H., Li, C., Evans, P., and de Bono, M. (2003). Inhibition of Caenorhabditis elegans social feeding by FMRFamide-related peptide activation of NPR-1. Nat. Neurosci. 6, 1178-1185. Abstract Article
Rojas, R.J., Yohe, M.E., Gershburg, S., Kawano, T., Kozasa, T., and Sondek, J. (2007). Gαq directly activates p63RhoGEF and Trio via a conserved extension of the Dbl homology-associated pleckstrin homology domain. J. Biol. Chem. 282, 29201-29210. Abstract Article
Rose, L. and Gönczy, P. Polarity establishment, asymmetric division and segregation of fate determinants in early C. elegans embryos (December 30, 2014) WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.30.2, http://www.wormbook.org. Abstract Article
Ross, E.M. (2014). G protein-coupled receptors: Multi-turnover GDP/GTP exchange catalysis on heterotrimeric G proteins. Cell. Logist. 4, e29391. Abstract Article
Sabatini, B.L., and Regehr, W.G. (1996). Timing of neurotransmission at fast synapses in the mammalian brain. Nature 384, 170-172. Abstract Article
Sánchez-Fernández, G., Cabezudo, S., García-Hoz, C., Benincá, C., Aragay, A.M., Mayor, F. Jr., and Ribas, C. (2014). Gαq signalling: the new and the old. Cell Signal. 26, 833-848. Abstract Article
Sanyal, S., Wintle, R.F., Kindt, K.S., Nuttley, W.M., Arvan, R., Fitzmaurice, P., Bigras, E., Merz, D.C., Hébert, T.E., van der Kooy, D., et al. (2004). Dopamine modulates the plasticity of mechanosensory responses in Caenorhabditis elegans. EMBO J. 23, 473-482. Abstract Article
Sawin, E.R., Ranganathan, R., and Horvitz, H.R. (2000). C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron 26, 619-631. Abstract Article
Schade, M.A., Reynolds, N.K., Dollins, C.M., and Miller, K.G. (2005). Mutations that rescue the paralysis of Caenorhabditis elegans ric-8 (synembryn) mutants activate the Gαs pathway and define a third major branch of the synaptic signaling network. Genetics 169, 631-649. Abstract Article
Schafer, W.F. (2006). Genetics of egg-laying in worms. Annu. Rev. Genet. 40, 487-509. Abstract Article
Schultheis, C., Brauner, M., Liewald, J.F., and Gottschalk, A. (2011). Optogenetic analysis of GABAB receptor signaling in Caenorhabditis elegans motor neurons. J. Neurophysiol. 106, 817-827. Abstract Article
Ségalat, L., Elkes, D.A., and Kaplan, J.M. (1995). Modulation of serotonin-controlled behaviors by Go in Caenorhabditis elegans. Science 267, 1648-1651. Abstract Article
Sellings, L., Pereira, S., Qian, C., Dixon-McDougall, T., Nowak, C., Zhao, B., Tyndale, R.F., and van der Kooy. D. (2013). Nicotine-motivated behavior in Caenorhabditis elegans requires the nicotinic acetylcholine receptor subunits acr-5 and acr-15. Eur. J. Neurosci. 37, 743-756. Abstract Article
Serrano-Saiz, E., Poole, R.J., Felton, T., Zhang, F., De La Cruz, E.D., and Hobert, O. (2013). Modular control of glutamatergic neuronal identity in C. elegans by distinct homeodomain proteins. Cell 155, 659-673. Abstract Article
Siderovski, D.P., Diversé-Pierluissi, M.A., and De Vries, L. (1999). The GoLoco motif: a Gαi/o binding motif and potential guanine-nucleotide exchange factor. Trends Biochem. Sci. 24, 340-341. Abstract Article
Sieburth, D., Ch’ng, Q., Dybbs, M., Tavazoie, M., Kennedy, S., Wang, D., Dupuy, D., Rual, J.F., Hill, D.E., Vidal, M., Ruvkun, G., and Kaplan, J.M. (2005) Systematic analysis of genes required for synapse structure and function. Nature 436, 510-517. Abstract Article
Sieburth, D., Madison, J.M., and Kaplan, J.M. (2007). PKC-1 regulates secretion of neuropeptides. Nat. Neurosci. 10, 49-57. Abstract Article
Siehler, S. (2009). Regulation of RhoGEF proteins by G12/13-coupled receptors. Br. J. Pharmacol. 158, 41-49. Abstract Article
Singh, V., and Aballay, A. (2012). Endoplasmic reticulum stress pathway required for immune homeostasis is neurally controlled by arrestin-1. J. Biol. Chem. 287, 33191-33197. Abstract Article
Song, B.M., and Avery, L. (2012). Serotonin activates overall feeding by activating two separate neural pathways in Caenorhabditis elegans. J. Neurosci. 32, 1920-1931. Abstract Article
Song, B.M., Faumont, S., Lockery, S., and Avery, L. (2013). Recognition of familiar food activates feeding via an endocrine serotonin signal in Caenorhabditis elegans. eLife 2, e00329. Abstract Article
Speese, S., Petrie, M., Schuske, K., Ailion, M., Ann, K., Iwasaki, K., Jorgensen, E.M., and Martin, T.F. (2007). UNC-31 (CAPS) is required for dense-core vesicle but not synaptic vesicle exocytosis in Caenorhabditis elegans. J. Neurosci. 27, 6150-6162. Abstract Article
Spencer, W.C., McWhirter, R., Miller, T., Strasbourger, P., Thompson, O., Hillier, L.W., Waterston, R.H., and Miller, D.M. III. (2014). Isolation of specific neurons from C. elegans larvae for gene expression profiling. PLoS One. 9, e112102. Abstract Article
Srinivasan, D.G., Fisk, R.M., Xu, H., and van den Heuvel, S. (2003). A complex of LIN-5 and GPR proteins regulates G protein signaling and spindle function in C. elegans. Genes Dev. 17, 1225-3129. Abstract Article
Srinivasan, S., Sadegh, L., Elle, I.C., Christensen, A.G., Faergeman, N.J., and Ashrafi, K. (2008). Serotonin regulates C. elegans fat and feeding through independent molecular mechanisms. Cell Metab. 7, 533-544. Abstract Article
Stawicki, T.M., Takayanagi-Kiya, S., Zhou, K., and Jin, Y. (2013). Neuropeptides function in a homeostatic manner to modulate excitation-inhibition imbalance in C. elegans. PLoS Genet. 9, e1003472. Abstract Article
Steger, K.A., and Avery, L. (2004). The GAR-3 muscarinic receptor cooperates with calcium signals to regulate muscle contraction in the Caenorhabditis elegans pharynx. Genetics 167, 633-463. Abstract Article
Sternweis, P.C., and Robishaw, J.D. (1984). Isolation of two proteins with high affinity for guanine nucleotides from membranes of bovine brain. J. Biol. Chem. 259, 13806-13813. Abstract Article
Sugiura, M., Fuke, S., Suo, S., Sasagawa, N., Van Tol, H.H., and Ishiura. S. (2005). Characterization of a novel D2-like dopamine receptor with a truncated splice variant and a D1-like dopamine receptor unique to invertebrates from Caenorhabditis elegans. J. Neurochem. 94, 1146-1157. Abstract Article
Suh, S.J., Park, Y.S., Lee, Y.S., Cho, T.J., Kaang, B.K., and Cho, N.J. (2001). Three functional isoforms of GAR-2, a Caenorhabditis elegans G-protein-linked acetylcholine receptor, are produced by alternative splicing. Biochem. Biophys. Res. Commun. 288, 1238-1243. Abstract Article
Sun, J., Liu, Y., and Aballay, A. (2012). Organismal regulation of XBP-1-mediated unfolded protein response during development and immune activation. EMBO Rep. 13, 855-860. Abstract Article
Sun, J., Singh, V., Kajino-Sakamoto, R., Aballay, A. (2011). Neuronal GPCR controls innate immunity by regulating noncanonical unfolded protein response genes. Science 332, 729-732. Abstract Article
Sunahara, R.K., Dessauer, C.W., and Gilman, A.G. (1996). Complexity and diversity of mammalian adenylyl cyclases. Annu. Rev. Pharmacol. Toxicol. 36, 461-480. Abstract Article
Suo, S., Culotti, J.G., and Van Tol, H.H. (2009). Dopamine counteracts octopamine signalling in a neural circuit mediating food response in C. elegans. EMBO J. 28, 2437-2448. Abstract Article
Suo, S., Kimura, Y., and Van Tol, H.H. (2006). Starvation induces cAMP response element-binding protein-dependent gene expression through octopamine-Gq signaling in Caenorhabditis elegans. J. Neurosci. 26, 10082-10090. Abstract Article
Suo, S., Sasagawa, N., and Ishiura, S. (2002). Identification of a dopamine receptor from Caenorhabditis elegans. Neurosci. Lett. 319, 13-16. Abstract Article
Suo, S., Sasagawa, N., and Ishiura, S. (2003). Cloning and characterization of a Caenorhabditis elegans D2-like dopamine receptor. J. Neurochem. 86, 869-878. Abstract Article
Sutherland, E.W. (1971). Studies on the Mechanism of Hormone Action. In Nobel Lectures, Physiology or Medicine 1971-1980, J. Lindsten, ed. (Signapore: World Scientific Publishing Co.), pp. 5-22. Abstract Article
Takesono, A., Cismowski, M.J., Ribas, C., Bernard, M., Chung, P., Hazard, S. III, Duzic, E., and Lanier, S.M. (1999) Receptor-independent activators of heterotrimeric G-protein signaling pathways. J. Biol. Chem. 274, 33202-33205. Abstract Article
Tall, G.G., Krumins, A.M., and Gilman, A.G. (2003). Mammalian Ric-8A (synembryn) is a heterotrimeric Gα protein guanine nucleotide exchange factor. J. Biol. Chem. 278, 8356-8362 Abstract Article
Tanis, J.E., Moresco, J.J., Lindquist, R.A., and Koelle, M.R. (2008). Regulation of serotonin biosynthesis by the G proteins Gαo and Gαq controls serotonin signaling in Caenorhabditis elegans. Genetics 178, 157-169. Abstract Article
Taussig, R., Tang, W.J., Hepler, J.R., and Gilman, A.G. (1994). Distinct patterns of bidirectional regulation of mammalian adenylyl cyclases. J. Biol. Chem. 269, 6093-6100. Abstract Article
Tesmer, J.J., Berman, D.M., Gilman, A.G., and Sprang, S.R. (1997). Structure of RGS4 bound to AlF4—activated Giα1: stabilization of the transition state for GTP hydrolysis. Cell 89, 251-261. Abstract Article
Tharmalingam, S., Burns, A.R., Roy, P.J., and Hampson, D.R. Orthosteric and allosteric drug binding sites in the Caenorhabditis elegans mgl-2 metabotropic glutamate receptor. Neuropharmacology 63, 667-674. Abstract Article
Thomas, C.J., Tall, G.G., Adhikari, A., and Sprang, S.R. (2008). Ric-8A catalyzes guanine nucleotide exchange on Gαi1 bound to the GPR/GoLoco exchange inhibitor AGS3. J. Biol. Chem. 283, 23150-23160. Abstract Article
Topper, S.M., Aguilar, S.C., Topper, V.Y., Elbel, E., and Pierce-Shimomura, J.T. (2014). Alcohol disinhibition of behaviors in C. elegans. PLoS One 9, e92965. Abstract Article
Trent, C., Tsung, N., and Horvitz, H.R. (1983). Egg-laying defective mutants of the nematode Caenorhabditis elegans. Genetics 104, 619-647. Abstract Article
Trudeau, L.E., Emery, D.G., and Haydon, P.G. (1996). Direct modulation of the secretory machinery underlies PKA-dependent synaptic facilitation in hippocampal neurons. Neuron 17, 789-797. Abstract Article
Tsalik, E.L., Niacaris, T., Wenick, A.S., Pau, K., Avery, L., and Hobert, O. (2003). LIM homeobox gene-dependent expression of biogenic amine receptors in restricted regions of the C. elegans nervous system. Dev. Biol. 263, 81-102. Abstract Article
Vaidya, A., Jain, S., Jain, A.K., Agrawal, A., Kashaw, S.K., Jain, S.K., and Agrawal R.K. (2013). Metabotropic glutamate receptors: a review on prospectives and therapeutic aspects. Mini Rev. Med. Chem. 13, 1967-1981. Abstract Article
Vashlishan, A.B., Madison, J.M., Dybbs, M., Bai, J., Sieburth, D., Ch’ng, Q., Tavazoie, M., and Kaplan, J.M. (2008). An RNAi screen identifies genes that regulate GABA synapses. Neuron 58, 346-361. Abstract Article
van der Linden, A.M., Moorman, C., Cuppen, E., Korswagen, H.C., and Plasterk, R.H. (2003). Hyperactivation of the G12-mediated signaling pathway in Caenorhabditis elegans induces a developmental growth arrest via protein kinase C. Curr Biol. 13, 516- 521. Abstract Article
van der Linden, A.M., Simmer, F., Cuppen, E., Plasterk, R.H. (2001). The G-protein β-subunit GPB-2 in Caenorhabditis elegans regulates the Goα-Gqα signaling network through interactions with the regulator of G-protein signaling proteins EGL-10 and EAT-16. Genetics 158, 221-235. Abstract Article
Vidal-Gadea, A., Topper, S., Young, L., Crisp, A., Kressin, L., Elbel, E., Maples, T., Brauner, M., Erbguth, K., Axelrod, A., et al. (2011). Caenorhabditis elegans selects distinct crawling and swimming gaits via dopamine and serotonin. Proc. Natl. Acad. Sci. U. S. A. 108, 17504-17509. Abstract Article
Wall, M.A., Coleman, D.E., Lee, E., Iñiguez-Lluhi, J.A., Posner, B.A., Gilman, A.G., and Sprang. S.R. (1995). The structure of the G protein heterotrimer Giα1β1γ2. Cell 83, 1047-1058. Abstract Article
Wang, D., Yu, Y., Li, Y., Wang, Y., and Wang, D. (2014). Dopamine receptors antagonistically regulate behavioral choice between conflicting alternatives in C. elegans. PLoS One 9, e115985. Abstract Article
Wang, H., Girskis, K., Janssen, T., Chan, J.P., Dasgupta, K., Knowles, J.A., Schoofs, L., and Sieburth, D. (2013). Neuropeptide secreted from a pacemaker activates neurons to control a rhythmic behavior. Curr. Biol. 23, 746-754. Abstract Article
Wang, Y., Tang, L., Feng, X., Du, W., and Liu, B.F. (2011). Ethanol interferes with gustatory plasticity in Caenorhabditis elegans. Neurosci. Res. 71, 341-347. Abstract Article
Wedegaertner, P.B., Wilson, P.T., and Bourne, H.R. (1995). Lipid modifications of trimeric G proteins. J. Biol. Chem. 270, 503-506. Abstract Article
White, J.G., Southgate, E., Thomson, J.N., and Brenner, S. (1986). The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 314, 1-340. Abstract Article
Whiteway, M., Hougan, L., Dignard, D., Thomas, D.Y., Bell, L., Saari, G.C., Grant, F.J., O’Hara, P., and MacKay, V.L. (1989). The STE4 and STE18 genes of yeast encode potential β and γ subunits of the mating factor receptor-coupled G protein. Cell 56, 467-477. Abstract Article
Wierda, K.D., Toonen, R.F., de Wit, H., Brussaard, A.B., and Verhage, M. (2007). Interdependence of PKC-dependent and PKC-independent pathways for presynaptic plasticity. Neuron 54, 275-290. Abstract Article
Williams, S.L., Lutz, S., Charlie, N.K., Vettel, C., Ailion, M., Coco, C., Tesmer, J.J., Jorgensen, E.M., Wieland, T., and Miller, K.G. (2007). Trio’s Rho-specific GEF domain is the missing Gαq effector in C. elegans. Genes Dev. 21, 2731-2746. Abstract Article
Wragg, R.T., Hapiak, V., Miller, S.B., Harris, G.P., Gray, J., Komuniecki, P.R., and Komuniecki, R.W. (2007). Tyramine and octopamine independently inhibit serotonin-stimulated aversive behaviors in Caenorhabditis elegans through two novel amine receptors. J. Neurosci. 27, 13402-13412. Abstract Article
Xiao, H., Hapiak, V.M., Smith, K.A., Lin, L., Hobson, R.J., Plenefisch, J., and Komuniecki, R. (2006). SER-1, a Caenorhabditis elegans 5-HT2-like receptor, and a multi-PDZ domain containing protein (MPZ-1) interact in vulval muscle to facilitateserotonin-stimulated egg-laying. Dev. Biol. 298, 379-391. Abstract Article
Yau, D.M., Yokoyama, N., Goshima, Y., Siddiqui, Z.K., Siddiqui, S.S., and Kozasa, T. (2003). Identification and molecular characterization of the Gα12-Rho guanine nucleotide exchange factor pathway in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 100, 14748-1453. Abstract Article
Yoshida, M., Oami, E., Wang, M., Ishiura, S., and Suo, S. (2014). Nonredundant function of two highly homologous octopamine receptors in food-deprivation-mediated signaling in Caenorhabditis elegans. J. Neurosci. Res. 92, 671-678. Abstract Article
Zhou, K.M., Dong, Y.M., Ge, Q., Zhu, D., Zhou, W., Lin, X.G,, Liang, T., Wu, Z.X., and Xu, T. (2007). PKA activation bypasses the requirement for UNC-31 in the docking of dense core vesicles from C. elegans neurons. Neuron 56, 657-669. Abstract Article
Zimmerman, G., and Soreq, H. (2006). Termination and beyond: acetylcholinesterase as a modulator of synaptic transmission. Cell Tissue Res. 326, 655-669. Abstract Article
*Edited by Iva Greenwald Last revised February 4, 2016. Published in its final form December 11, 2018. This chapter should be cited as: Koelle M.R. Neurotransmitter signaling through heterotrimeric G proteins: insights from studies in C. elegans. (December 11, 2018), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.75.2, http://www.wormbook.org.
Copyright: © 2016 Michael R. Koelle. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. Email: [email protected]
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.