Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
Genetic suppression has provided a very powerful tool for analyzing C. elegans. Suppression experiments are facilitated by the ability to handle very large numbers of individuals and to apply powerful selections. Because the animal grows as a self-fertilizing diploid, both dominant and recessive suppressors can be recovered. Many different kinds of suppression have been reported. These are discussed by category, with examples, together with discussion of how suppressors can be used to interpret the underlying biology, and to enable further experimentation. Suppression phenomena can be divided into intragenic and extragenic classes, depending on whether the suppressor lies in the same gene as the starting mutation, or in a different gene. Intragenic types include same-site replacement, compensatory mutation, alteration in splicing, and reversion of dominant mutations by cis-knockout. Extragenic suppression can occur by a variety of informational mechanisms, such as alterations in splicing, translation or nonsense-mediated decay. In addition, extragenic suppression can occur by bypass, dosage effects, product interaction, or removal of toxic products. Within signaling pathways, suppression can occur by modulating the strength of signal transmission, or by epistatic interactions that can reveal the underlying regulatory hierarchies. In C. elegans biology, the processes of muscle development, vulva formation and sex determination have provided remarkably rich arenas for the investigation and exploitation of suppression.
Genetic suppressors provide some of the most powerful tools available for exploring gene expression, function and interaction. A suppressor mutation can be defined as a second mutation, at a site distinct from the mutation under study, that suppresses the phenotypic effect of the first mutation. C. elegans is especially suited for suppression analysis, for several practical reasons:
Very large numbers of individuals can be handled, so that extremely rare mutational events can occur within a single experimental population.
Strong selections can be applied, which may permit the detection and recovery of a single individual from a population of one hundred thousand or more.
Mutations are easily generated, particularly by the chemical mutagen EMS.
The predominant mode of reproduction is by self-fertilization, so that both recessive and dominant suppressors can be recovered readily.
Genetic suppression can arise in a variety of ways, which are described in the sections below. Depending on the nature of the suppressive interaction, different kinds of information can be obtained about the initial gene studied.
In the simplest and least informative scenario, if a mutant carries a single base change difference from wildtype, then reversion can occur simply by mutating the sequence back to wildtype. However, intracodonic suppression can also occur, if a different base change can result in a functional coding sequence. An example is provided by a Gln-to-Pro mutant of dpy-31, encoding a collagen-processing protease, which has been repeatedly reverted either by back mutation, Pro-to-Gln (CCT -> CTT), or by the only other acceptable change in this codon, Pro-to-Ser (CCT -> TCT) (Novelli et al., 2004).
In principle, missense mutations that result in a non-functional protein can sometimes be suppressed by introducing another change elsewhere in the protein. Relatively few cases of this effect have been studied so far in C. elegans. An example is provided by the reversion of temperature-sensitive missense mutations in glp-1, which encodes a cell interaction protein; this experiment yielded a variety of second-site missense changes that are able to correct the original defect (Lissemore et al., 1993).
Sometimes intragenic reversion can be achieved by a second mutation that leads to omission of a deleterious mutation by means of exon skipping. This is particularly likely to happen with genes that produce multiple splice variants, as illustrated by studies of unc-52. This gene encodes multiple isoforms of perlecan, which are proteins required for myofilament anchorage to the basement membrane underlying muscle cells. Nonsense mutations located in some of the alternatively spliced exons can be suppressed by a G-to-A change in the acceptor site adjoining the mutant exon, which leads to its omission; the resulting RNA remains in frame and produces a functional protein (Rogalski et al., 1995).
Most genes in C. elegans are haplo-sufficient, so the majority of dominant mutations are associated with gain-of-function effects, which result in a toxic or inappropriately expressed gene product (Park and Horvitz, 1986). The dominant effect of such a mutation can be suppressed by introducing a second mutation elsewhere in the mutated gene that results in partial or complete loss-of-function in its product.
An early example was provided by studies of the myosin gene unc-54. Some dominant mutations of unc-54 cause slow movement or paralysis in single dose, by poisoning thick filament assembly. These mutations, such as the allele e1152, can be reverted by introducing null or partial loss-of-function mutations in cis. Heterozygotes carrying just the dominant allele (e1152/+) are slow, whereas e1152 null / + heterozygotes are wildtype. The unc-54 gene is essential for movement, so e1152 null double homozygotes are paralysed, but the mutations are initially recovered in healthy heterozygotes with normal movement. Selection for improved movement of e1152/+ heterozygotes therefore provided an efficient means of recovering many mutations in this gene (Anderson and Brenner, 1984).
Similar strategies have been used frequently and effectively to generate multiple loss-of-function mutations in important or essential genes.
Suppressing the effects of a mutant gene by changes elsewhere in the genome can occur in a number of ways. Extragenic suppression is usually revealed by crossing a revertant line with wildtype, and observing the re-appearance of the original mutant after Mendelian segregation. Numerous cases of extragenic suppression have now been identified and analysed in greater or lesser depth. Many different phenomena are involved in the various cases. About 40 genes in the sup class have been defined thus far, and in addition separate gene classes have been established for many of the more specific processes that have been studied. As shown in Table 1, research on 21 suppressor gene classes has been published. At least 20 further classes have been established (see Wormbase) but their properties are as yet unpublished. It should be noted that suppressor mutations frequently turn out to be special alleles of genes that have already been well studied and given more informative names, so the names of suppressor loci are more transient than most.
Table 1. Suppressor gene classes
| Gene name | Explanation | Initial references |
|---|---|---|
| cps | CED-3 Protease Suppressor | Wang et al., 2002 |
| des | DEgeneration Suppressor | Treinin et al., 1998 |
| eor | Egl-1 suppressor/DiO uptake defect/Raf enhancer | Howard and Sundaram, 2002 |
| ksr | Kinase Suppressor of activated Ras | Sundaram and Han, 1995; Kornfeld et al., 1995 |
| scd | Suppressor of Constitutive Dauer formation | Inoue and Thomas, 2000 |
| sel | Suppressor/Enhancer of Lin-12 | Sundaram and Greenwald, 1993 |
| seu | Suppressor of Ectopic Unc-5 | Colavita and Culotti, 1998 |
| sgs | Suppressor of G protein S defect | Korswagen et al., 1998 |
| sli | Suppressor of LIneage defect | Jongeward et al., 1995 |
| smg | Suppressor, Morphological effect on Genitalia | Hodgkin et al., 1989 |
| smu | Suppressor of Mec and Unc defects | Lundquist and Herman, 1994 |
| soc | Suppressor Of Clr | Selfors et al., 1998 |
| sog | Suppressor Of Glp | Maine and Kimble, 1993 |
| sol | Suppressor Of Lurcher movement defect | Zheng et al., 2004 |
| sop | Suppressor Of Pal | Zhang and Emmons, 2000 |
| spr | Suppressor of PResenilin defect | Wen et al. 2000 |
| srl | Suppressor of Roller Lethality | Barbazuk et al., 1994 |
| sup | SUPpressor | Riddle and Brenner, 1978; Waterston and Brenner, 1978 |
| sur | SUppressor of activated Ras | Lackner et al., 1994; Wu and Han, 1994 |
| sus | SUppressor of Suppressor | Brown and Riddle, 1985 |
| suv | SUppressor of Vulvaless | Hajnal et al., 1997 |
Space does not permit a full description of all the classes listed in Table 1, but some of the major types of suppression that have been encountered are discussed below, using particular examples.
Alteration of the general machinery for transcription, RNA processing and translation can sometimes ameliorate or suppress certain kinds of mutation in many different genes. The hallmarks of informational suppression, as this phenomenon is known, are allele-specificity and gene-non-specificity. Three kinds of informational suppression have been described so far in C. elegans: nonsense suppression; suppression by modified splicing; suppression by loss of nonsense-mediated decay (NMD).
A large fraction of point mutations identified in C. elegans are changes from sense to nonsense codons, either UAG (amber), UAA (ochre) or UGA (opal), resulting in polypeptide chain termination and (usually) complete or almost complete loss of gene function. These nonsense mutants can potentially be suppressed by nonsense suppressor tRNAs, in which a sense codon has been changed to be able to decode the nonsense triplet. In practice, only amber suppressors have been recovered. Early work identified two suppressor loci, sup-5 and sup-7, which could act to suppress certain mutations in a wide variety of genes (Waterston and Brenner, 1978; Waterston, 1981). These were subsequently shown to be amber suppressor mutations of two Trp tRNA genes, carrying alterations in their anticodons from CCA to CTA (Wills, et al., 1983; Bolten et al., 1984).
There are twelve members of the Trp tRNA family in C. elegans, but only two can be mutated to yield strong amber suppressors. The same two mutations, sup-5 and sup-7, were isolated repeatedly in independent experiments (Waterston, 1981). Weak amber suppressors were subsequently obtained for six additional members of the set (see Table 2), by selecting for suppressors using amber mutants that are very easily suppressed such as several in the sex determination gene tra-3 (Hodgkin, 1985; Kondo et al., 1988; Kondo et al., 1990). Probably only a trace of full length TRA-3 protein is sufficient to provide enough activity for normal sexual development, so weak suppressors can be recovered. As indicated in Table 2, the eight identified amber suppressors appear to be expressed at different levels, and they also exhibit some differences between tissues in the strength of suppression (Kondo et al., 1990). In vitro observations on the transcription of these genes that are consistent with these different strengths have been reported (Li et al, 1988). The differences in strength show that flanking sequences can exert a strong influence on expression level of these tRNAs, despite the fact that all eight are identical in the internal promoter sequences believed to be sufficient for Pol III transcription of tRNA genes.
Table 2. Amber nonsense suppressors
| Gene name (allele) | Class (other name) | Approximate relative strength | Reference |
|---|---|---|---|
| sup-5(e1464) | Trp (rtw-1) | 7 | Waterston and Brenner (1978) |
| sup-7(st5) | Trp (rtw-2) | 8 | Waterston (1981) |
| sup-21(e1957) | Trp (rtw-10) | 3 | Hodgkin (1985) |
| sup-24(st354) | Trp (rtw-4) | 4 | Kondo et al. (1988) |
| sup-28(e2058) | Trp (rtw-8) | 2 | Kondo et al. (1988) |
| sup-29(e1986) | Trp (rtw-9) | 1 | Kondo et al. (1988) |
| sup-33(st389) | Trp (rtw-11) | 4 | Kondo et al. (1990) |
| sup-34(e2227) | Trp (rtw-12) | 3 | Kondo et al. (1990) |
| sup-22(e2057) | Non-Trp | 2 | Hodgkin (1985) |
| sup-23(e2059) | ?Non-Trp | 2 | Hodgkin (1985) |
| sup-32(e1958) | Non-Trp | 2 | Hodgkin (1985) |
| sup-?(st402) | Non-Trp | 3 | Kondo et al. (1990) |
| sup-?(st414) | Non-Trp | 2 | Kondo et al. (1990) |
Too much suppression is clearly deleterious, because animals carrying two doses of sup-7 or sup-5 are inviable or subviable, especially at low temperature (Waterston, 1981). Presumably the deleterious effects result from too much readthrough of natural stop codons. Selection for improved low-temperature viability of sup-7 animals was used to generate deletions or loss-of-function mutations of sup-7, suggesting that the inviability is due to readthrough rather than loss of the normal function of the gene. The same effect may explain why ochre or opal suppressors have not been recovered.
Additional weak amber suppressors have been isolated, which do not correspond to Trp tRNA genes, but they have not yet been identified at the molecular level (Table 2). Some of these are likely to be Gln tRNA genes, because a Gln CTG anticodon can be readily mutated to read amber codons. Suppression by an amber-suppressing Ser tRNA from Drosophila, introduced as a transgene, has been reported (Pilgrim and Bell, 1993), demonstrating that endogenous Ser tRNA genes might also yield amber suppressors. As well as providing valuable tools for examining mutations in particular genes and for studying tRNA gene expression, amber suppressors have been useful in the development of methods for microinjection and transformation of C. elegans (Kimble et al., 1982; Fire, 1986) The dose-sensitivity of strong amber suppressors also allows selection of single-copy transformants (Fire et al., 1990).
Two kinds of suppression by modified splicing have been reported. The first kind depends on compensatory mutations in splice-site recognition machinery, and the second is associated with mutations that affect alternative splicing.
The first kind involves allele-specific suppression of mutations that change the conserved and essential GU donor sequence at the 5’ end of an intron, by means of compensatory changes in the sequence of a U1 snRNA gene. In the case of sup-6, a donor site mutation (AGgu --> AGau) in allele e309 of unc-13 is suppressed by a change in one of the U1 snRNA genes (…UCCAUU… --> UCUAUU) (Zahler et al., 2004). In a related case, sup-39, a donor site mutation (AGgu --> AGuu) in allele e936 of unc-73 is suppressed by a change in a different U1 snRNA gene (…UCCAUU… -->… UCAAUU…) (Run et al., 1996; Roller et al., 2000; Zahler et al., 2004). These effects are consistent with the proposed role of U1 in pairing at the donor site during splicing.
In C. elegans, there are 12 genes encoding U1 snRNA, yet only two have yielded suppressors of this type (Zahler et al., 2004). As with the failure to find ochre or opal suppressors discussed above, it is possible that most U1 suppressor mutations will be either too weak, because of redundancy, or too deleterious, because of effects on normal genes. In support of the latter possibility, both sup-6 and sup-39 exhibit defects when homozygous: sup-6 homozygotes arrest in late-larval development, and 50% of sup-39 homozygotes die as embryos. In addition, sup-39 animals that escape this embryonic lethality frequently exhibit two rows of oocytes rather than one in adult hermaphrodites, an unusual and unexplained phenotype (Run et al., 1996).
The second kind of extragenic suppression by altered splicing involves the regulation of alternative splicing. As described above, some mutations of the unc-52 perlecan gene are located in alternatively spliced exons and can be suppressed by intragenic mutations that cause exon-skipping. A subset of these, those located in exon 17, can also be suppressed by loss-of-function mutations in two genes, smu-1 and smu-2, which encode proteins homologous to mammalian spliceosome-associated factors (Spartz et al., 2004). Loss of these factors leads to enhanced skipping of exon 17, and consequently to suppression of mutations affecting this exon. The smu-1 and smu-2 mutations also weakly suppress the pleiotropic effects of mutations in mec-8, which encodes another regulator of alternative splicing (Lundquist and Herman, 1994). This suggests that the SMU-1 and SMU-2 factors can affect the splicing of multiple genes, although neither is essential for viability. It seems very likely that further cases of suppression by modified splicing will be encountered in the future.
Mutations resulting in a premature stop codon trigger the NMD system of message degradation. If the NMD system is absent, then mRNAs with such codons have longer lifetimes, increasing the chance of readthrough at the stop codon and the production of a functional protein. Alternatively, if the stop codon is located close to the end of the message, the truncated polypeptide produced after translation of the stabilized message may be long enough to have substantial activity. Nonsense mutations are frequently generated in C. elegans forward mutagenesis screens, so a variety of suppressor screens have yielded mutations in the C. elegans NMD genes (Hodgkin et al., 1989). Seven genes required for NMD have been identified in this way, which are called smg (for Suppressor with Morphogenetic effect on Genitalia). Loss of NMD does not lead to inviability in C. elegans, but does result in an abnormal morphology of the hermaphrodite vulva and of the male tail, hence the name. Suppression is generally weaker than with amber suppression.
Some cases of suppression probably result from general effects on the physiology of the animal, which can alter the expression of a variety of different genes. An example is provided by the recovery of mutations in eight or more cuticle collagen genes, all of which cause a ‘dumpy left roller’ phenotype, as weak suppressors of two quite different mutant defects: germline proliferation, in the case of glp-1 (Maine and Kimble, 1989), and embryonic gastrulation, in the case of emb-5 (Nishiwaki and Miwa, 1998). The mechanism involved is unknown, but indirect effects via alteration of extracellular matrix or of stress response have been suggested as possible explanations.
Sometimes the lack of a particular protein can be compensated for by increasing the amount of an alternative, related protein. This is illustrated by the analysis of a gene initially called sup-3 (Riddle and Brenner, 1978), identified by mutations that acted to suppress unc-54 or unc-15 mutants; these lack either the major body wall myosin (UNC-54) or another thick filament component, paramyosin (UNC-15). Molecular analysis of sup-3 showed that its mutant alleles are duplications or triplications of myo-3, which encodes a minor myosin. Normally MYO-3 accounts for only 15% of the body wall myosin, but its level is increased in the duplication alleles, and can thereby compensate for the lack of UNC-54 or UNC-15 (Maruyama et al., 1989).
Suppression by increased dosage can be regarded as a special case of bypass suppression, wherein an alternative protein or pathway is up-regulated or de-regulated to remove the need for a defective component. Mutations that activate alternative pathways are often encountered as a result of suppressor analysis. Screens for suppressors of pal-1, the C. elegans homolog of the developmental regulator caudal, illustrate this effect (Zhang and Emmons, 2000). In normal development, pal-1 expression in certain tail cells depends on a positive regulatory element located in one of its introns. A screen was carried out for suppressors of a pal-1 mutation affecting this intronic element, which led to the identification of three sop (Suppressor Of Pal) genes. These encode factors that act as general repressors of an alternative mode of pal-1 transcription. Two of the sop genes encode components of the transcriptional Mediator complex. Loss of these factors results in expression of pal-1 in the tail cells, and hence to suppression by bypassing the need for the intronic regulatory element.
An abnormal protein can sometimes be restored to greater functionality by changes in an interacting protein. Suppression by such a mechanism is usually allele-specific, and provides in vivo evidence for protein interaction. An example is provided by mutations of the muscle protein gene unc-22, which cause a ‘twitcher’ abnormal movement phenotype. Some revertants of certain unc-22 alleles were found to be unusual missense alleles of the body wall myosin gene unc-54, suggesting that the UNC-22 and UNC-54 proteins interact directly (Moerman et al., 1982). Both these proteins are, indeed, located in the thick filaments of the myofilament lattice (Sarcomere assembly in C. elegans muscle).
Another example is provided by suppression of certain dominant mutations in unc-105, a gene encoding a putative mechanosensory channel in muscle cells, by a specific missense alteration in let-2, which encodes a basement membrane collagen (Liu et al., 1996). A plausible interpretation is that UNC-105 and LET-2 interact directly, providing anchorage for the sensory channel and allowing it to act as a stretch sensor.
A situation sometimes encountered in C. elegans genetics is the appearance of an abnormal phenotype as the result of a neomorphic change in a gene, resulting in the production of a toxic protein. Such mutations are usually but not always dominant (sometimes they are recessive because in heterozygotes the abnormal protein is outcompeted by normal protein). Suppression studies have been carried out with such mutations in a variety of different genes. Any alteration that removes the toxic protein, or prevents it from acting, will behave as a suppressor.
The simplest situation involves the introduction of a second inactivating mutation in cis to the dominant allele. This is exemplified by dominant mutations of the myosin gene unc-54, as already described.
A more complex situation was encountered with the gene unc-93, which has been subjected to extensive suppression analysis. A rare mutation of unc-93, e1500, causes a distinctive movement and egg-laying defect referred to as the ‘rubber-band’ phenotype. This phenotype is almost completely recessive, although genetic tests indicate that e1500 is neomorphic in character. Null mutations introduced in cis to e1500 result in normally moving animals, when homozygous. Therefore, the unc-93 gene is dispensable, but can be mutated to generate a product that is able to poison muscle contraction (Greenwald and Horvitz, 1980). The e1500 phenotype can also be suppressed by null mutations in three unlinked genes, sup-9, sup-10 and sup-18, which act as recessive suppressors, as well as by specific dominant mutations of another gene, sup-11 (Greenwald and Horvitz, 1982). Moreover, ‘rubber-band’ alleles of sup-9 and sup-10 were subsequently found, which can be suppressed by null alleles of unc-93 (Greenwald and Horvitz, 1986; Levin and Horvitz, 1992). Molecular analysis indicates that these genes encode components of a muscle-specific potassium channel: the SUP-9 sequence reveals that it belongs to the family of two-pore K+ channel proteins, and SUP-10 and UNC-93 are membrane proteins that co-localize with SUP-9 in muscle cells (Levin and Horvitz, 1993; de la Cruz et al., 2003). The current interpretation is therefore that this multi-protein channel complex is dispensable for normal muscle function, but it can be mutated so as to interfere severely with contraction, presumably by affecting K+ flux. The other two interacting genes, sup-11 and sup-18, likely encode additional components, but sup-11 is known to be an essential gene, in contrast to the other four (because null mutations of sup-11 are embryonic lethal), so it must be involved in other processes as well.
A comparable situation was found in the case of the deg-3 gene, which encodes a nicotinic acetylcholine receptor subunit. Rare dominant alleles of deg-3 cause neuronal degeneration (hence the name), but the degeneration can be suppressed by loss-of-function mutations of des-2 (DEgeneration Suppressor). The des-2 gene was found to encode another receptor subunit, and co-expression of wildtype DEG-3 and DES-2 in Xenopus oocytes resulted in formation of a functional acetylcholine-gated channel. Co-expression of mutant DEG-3 with wildtype DES-2 resulted in an abnormal channel. Expression of either subunit alone, however, does not allow formation of a channel, thereby explaining the mechanism of suppression (Treinin et al., 1998).
The efficiency of a signal transduction pathway is usually modulated by interacting components that can increase or decrease the strength of a signal. This is especially true in multicellular organisms, where the same pathway may be used for multiple purposes in different tissues, and can be fine-tuned by these modulators so as to allow optimal function in each context. The presence of modulators is often revealed by suppression analysis. Good illustrations of this type of suppression are provided by extensive work on the LIN-12/NOTCH receptor, which is involved in numerous important signaling events during development (see LIN-12/Notch signaling in C. elegans). Suppression experiments have been carried out using both hypomorphic, partial loss-of-function alleles (Sundaram and Greenwald, 1993) and hypermorphic, gain-of-function alleles of lin-12 (Levitan and Greenwald, 1995; Tax et al., 1997). The first kind of experiment yielded mutations in negative regulators of LIN-12 activity, such as sel-1, encoding an extracellular protein (Grant and Greenwald, 1996), and sel-10, encoding an F-box protein predicted to be involved in ubiquitin-mediated turnover of LIN-12 (Hubbard et al., 1997). SEL-10 was independently identified as a factor affecting sex determination, again partly as a result of suppressor analysis (Jager et al., 2004).
The second kind of experiment, suppressing dominant gain-of-function alleles of lin-12, generated mutations in positive regulators. These included SUP-17, a protein belonging to the ADAM protease family, which perhaps acts by cleaving and activating the extracellular domain of the LIN-12 receptor (Wen et al., 1997). Also identified were SEL-5, a serine/threonine kinase (Fares and Greenwald, 1999) and SEL-12, which is related to mammalian presenilin proteins and therefore of great medical interest (Levitan and Greenwald, 1995). Not surprisingly, sel-12 mutations have an egg-laying defect in a lin-12(+) background; this in turn has allowed further suppressor screens for mutations correcting this defect, and the identification of spr (Suppressor of Presenilin) genes (Wen et al. 2000).
Suppression analysis has been invaluable in unraveling regulatory pathways in C. elegans. Many important events in development depend on signal transduction events, whereby an inductive signal emanating from one cell leads to a transcriptional response in target cells or tissues. The transduction of the signal involves a chain of interactions, with receptors, second messengers and transcription factors, and the interactions may be either positive or negative at each step. Mutations that decrease or increase signaling at one step can be epistatically suppressed by antagonistic mutations acting at a downstream step, which in turn can be subjected to further analysis. If the pathway involves a linear series of interactions, then the epistatic relationships of mutations in the different genes can be used to deduce the order and nature of the steps involved. More detailed discussion of the rules and interpretation of epistasis can be found elsewhere (Avery and Wasserman 1992).
Two good examples are provided by the vulval induction pathway (see RTKRas/MAP kinase signaling and the sex determination pathway (see Somatic sex determination and Sex-determination in the germ line). In the first case, opposite mutant phenotypes result from either too little or too much activation of the ERK MAP kinase pathway in the vulval precursor cells. These phenotypes are called Vul (vulvaless, failure to induce or complete vulval differentation) and Muv (multivulva, excess vulval differentiation) respectively. Both kinds of phenotype are very conspicuous in adult hermaphrodites, and both are associated with egg-laying defects and reduced fertility, so suppressors are easily detected. Mutations that suppress a Vul phenotype often lead to a Muv phenotype on their own, and vice versa. For example, screens for suppressors of a loss-of-function Vul mutation in let-23, encoding the EGF receptor required for vulval induction, yielded a gain-of-function in let-60, encoding the downstream Ras protein in this signal transduction pathway. The let-60(gf) mutation has a Muv phenotype on its own, and screens for suppressors of the Muv phenotype led to the isolation of mutations in ten or more genes encoding further downstream components or modifiers of the pathway, most of which have a Vul phenotype on their own. This kind of analysis has led to the identification of thirty or more genes involved in EGF signaling, and has played a major role in the very detailed elucidation of its properties and function (see RTKRas/MAP kinase signaling). For a summary of the suppressor screens that have been carried out, see Sternberg and Han (1998).
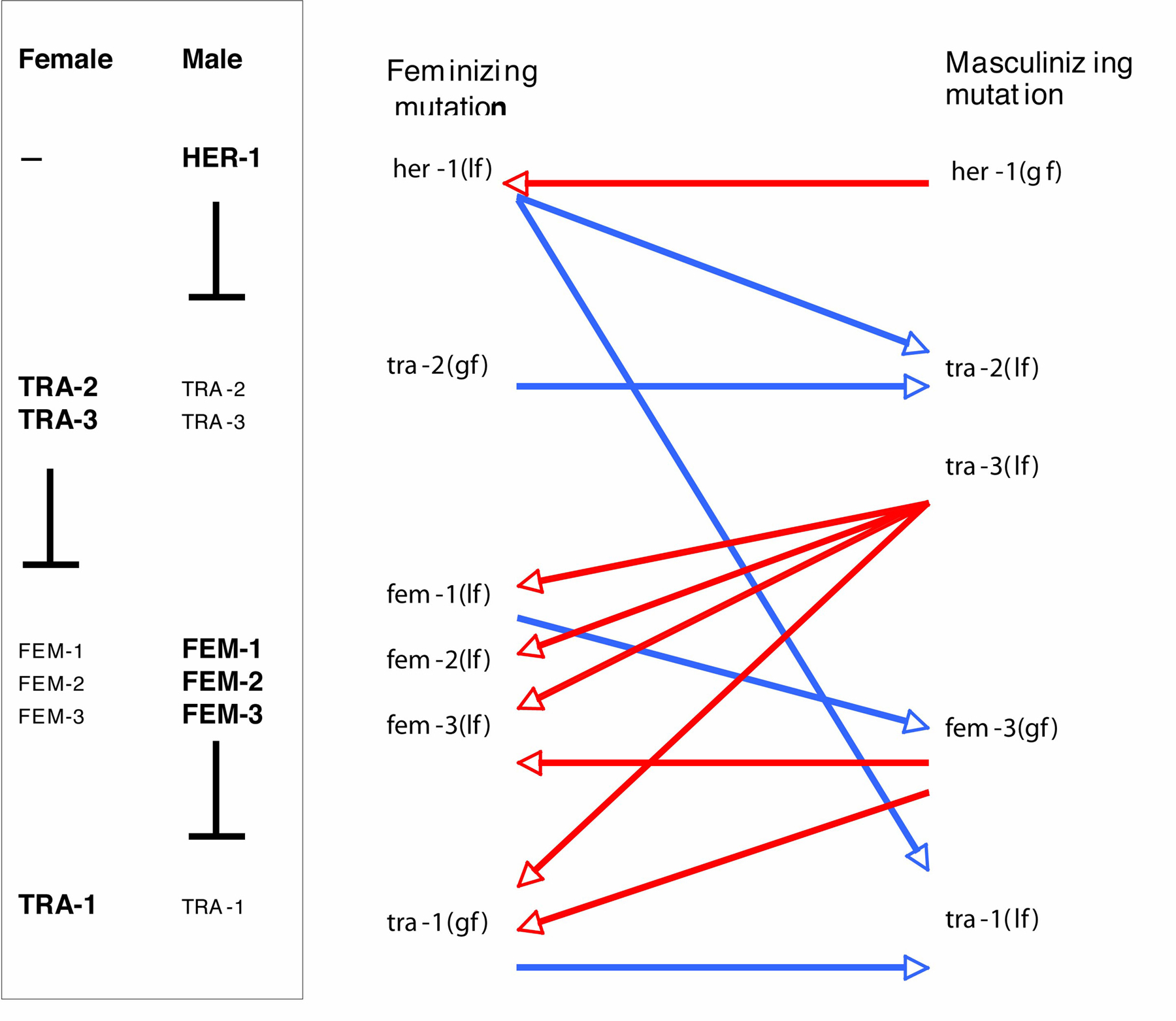
In the case of sex determination, the opposite phenotypes caused by mutation are Masculinized and Feminized. It is frequently possible (for example by use of temperature-sensitive mutants) to set up a situation in which a large population of worms develops only along one sexual path. Such a pure male or pure female population will yield no progeny, so there is extremely effective selection for any suppressor mutation that reverts or ameliorates the phenotype, and thereby allows cross- or self-fertilization, and the production of progeny. An extensive series of experiments of this type has been carried out in a number of different laboratories, as summarized in Figure 1. Both loss-of-function and gain-of-function mutations have been isolated at each of four regulatory steps, largely as a result of this approach. For example, selections for suppressors of the masculinizing mutations of tra-3 yielded feminizing (loss-of-function) alleles of fem-1, fem-2 and fem-3, as well as gain-of-function alleles of tra-1 (Hodgkin, 1986). Amber alleles of tra-3 were used in some of these selections, which consequently also yielded many nonsense suppressors and NMD (smg) suppressors (Hodgkin, 1985; Hodgkin, 1986).
|
Figure 1. Suppressor screens and selections for sex determination mutants. Part of the signal transduction cascade controlling sex in C. elegans is shown at left. In hermaphrodites, the feminizing transcription factor TRA-1 promotes female development, and the membrane receptor TRA-2 prevents inhibition of TRA-1 by the FEM proteins. In males, TRA-1 is prevented from acting by the FEM proteins, because TRA-2 action is blocked by binding the extracellular male-specific protein HER-1. At right are shown some of the suppressive interactions that led to this model. Red arrows indicate isolation of feminizing suppressors that counteract masculinization, and blue arrows indicate isolation of masculinizing suppressors that counteract feminization. For more detailed explanation, see text and chapters on sex determination.
Almost every known control pathway in C. elegans has been subjected to some kind of suppressor analysis, and the results have frequently been critical in elucidating properties and testing models. Genetic suppression must also happen naturally, and is certain to play an important role in evolution. Whether new kinds of suppression will become apparent, as further genes and processes are subjected to investigation, remains to be seen.
Anderson, P., and Brenner. S. (1984). A selection for myosin heavy-chain mutants in the nematode C. elegans. Proc. Natl. Acad. Sci. USA 81, 4470–4474. Abstract
Avery, L., and Wasserman, S. (1992). Ordering gene function: the interpretation of epistasis in regulatory hierarchies. Trends Genet. 8, 312–316. Abstract
Barbazuk, W.B., Johnsen, R.C., and Baillie, D.L. (1994). The generation and genetic analysis of suppressors of lethal mutations in the Caenorhabditis elegans rol-3(V) gene. Genetics 136, 129–143. Abstract
Bolten, S.L., Powell-Abel, P., Fischhoff, D.A., and Waterston, R.H. (1984). The sup-7(st5) X gene of Caenorhabditis elegans encodes a tRNATrpUAG amber suppressor. Proc. Natl. Acad. Sci. USA 81, 6784–6788. Abstract
Brown, S.J., and Riddle, D.L. (1985). Gene interactions affecting muscle organization in Caenorhabditis elegans. Genetics 110, 421–440. Abstract
Colavita, A., and Culotti, J.G. (1998). Suppressors of ectopic UNC-5 growth cone steering identify eight genes involved in axon guidance in Caenorhabditis elegans. Dev. Biol. 194, 72–85. Abstract Article
de la Cruz, I.P., Levin, J.Z., Cummins, C., Anderson, P., and Horvitz, H.R. (2003). sup-9, sup-10, and unc-93 may encode components of a two-pore K+ channel that coordinates muscle contraction in Caenorhabditis elegans. J. Neurosci. 23, 9133–9145. Abstract
Fares, H., and Greenwald, I. (1999). SEL-5, a serine/threonine kinase that facilitates lin-12 activity in Caenorhabditis elegans. Genetics 153, 1641–1654. Abstract
Fire A, Kondo K, Waterston R. (1990). Vectors for low copy transformation of C. elegans. Nucleic Acids Res. 18, 4269–4270. Abstract
Grant, B., and Greenwald, I. (1996). The Caenorhabditis elegans sel-1 gene, a negative regulator of lin-12 and glp-1, encodes a predicted extracellular protein. Genetics 143, 237–247. Abstract
Greenwald, I.S., and Horvitz, H.R. (1980). unc-93(e1500): a behavioral mutant of Caenorhabditis elegans that defines a gene with a wild-type null phenotype. Genetics 96, 147–164. Abstract
Greenwald, I.S., and Horvitz, H.R. (1982). Dominant suppressors of a muscle mutant define an essential gene of Caenorhabditis elegans. Genetics 101, 211–225. Abstract
Greenwald, I.S., and Horvitz, H.R. (1986). A visible allele of the muscle gene sup-10 X of C. elegans. Genetics 113, 63–72. Abstract
Hajnal, A., Whitfield, C.W., and Kim, S.K. (1997). Inhibition of Caenorhabditis elegans vulval induction by gap-1 and by let-23 receptor tyrosine kinase. Genes Dev. 11, 2715–2728. Abstract
Hodgkin, J. (1985). Novel nematode amber suppressors. Genetics 111, 287–310. Abstract
Hodgkin, J. (1986). Sex determination in the nematode C. elegans: analysis of suppressors and characterization of fem genes. Genetics 114, 15–52. Abstract
Hodgkin, J., Papp, A., Pulak, R., Ambros, V., and Anderson, P. (1989). A new kind of informational suppression in the nematode Caenorhabditis elegans. Genetics 123, 301–313. Abstract
Howard, R.M., and Sundaram, M.V. (2002). C. elegans EOR-1/PLZF and EOR-2 positively regulate Ras and Wnt signaling and function redundantly with LIN-25 and the SUR-2 mediator component. Genes Dev. 16, 1815–1827. Abstract Article
Hubbard, E.J., Wu, G., Kitajewski, J., and Greenwald, I. (1997). sel-10, a negative regulator of lin-12 activity in Caenorhabditis elegans, encodes a member of the CDC4 family of proteins. Genes Dev. 11, 3182–3193. Abstract
Inoue, T., and Thomas, J.H. (2000). Suppressors of transforming growth factor-β pathway mutants in the Caenorhabditis elegans dauer formation pathway. Genetics 156, 1035–1046. Abstract
Jager, S., Schwartz, H.T., Horvitz, H.R., and Conradt, B. (2004). The Caenorhabditis elegans F-box protein SEL-10 promotes female development and may target FEM-1 and FEM-3 for degradation by the proteasome. Proc. Natl. Acad. Sci. USA 101, 12549–12554. Abstract Article
Jongeward, G.D., Clandinin, T.R, and Sternberg, P.W. (1995). sli-1, a negative regulator of let-23-mediated signaling in C. elegans. Genetics 139, 1553–1566. Abstract
Kimble, J., Hodgkin, J., Smith, T., Smith, J. (1982). Suppression of an amber mutation by microinjection of suppressor tRNA in C. elegans. Nature 299, 456–458. Abstract Article
Kondo, K., Hodgkin, J., and Waterston, R.H. (1988). Differential expression of five tRNA(UAGTrp) amber suppressors in Caenorhabditis elegans. Mol. Cell Biol. 8, 3627–3635. Abstract
Kondo, K., Makovec, B., Waterston, R.H., and Hodgkin, J. (1990). Genetic and molecular analysis of eight tRNA(Trp) amber suppressors in Caenorhabditis elegans. J. Mol. Biol. 215, 7–19. Abstract
Kornfeld, K., Hom, D.B., and Horvitz, H.R. (1995). The ksr-1 gene encodes a novel protein kinase involved in Ras-mediated signaling in C. elegans. Cell 83, 9039–9013. Abstract Article
Korswagen, H.C., van der Linden, A.M., and Plasterk, R.H. (1998). G protein hyperactivation of the Caenorhabditis elegans adenylyl cyclase SGS-1 induces neuronal degeneration. EMBO J. 17, 5059–5065. Abstract Article
Lackner, M.R., Kornfeld, K., Miller, L.M., Horvitz, H.R., and Kim, S.K. (1994). A MAP kinase homolog, mpk-1, is involved in ras-mediated induction of vulval cell fates in Caenorhabditis elegans. Genes Dev. 8, 160–173. Abstract
Levin, J.Z., and Horvitz, H.R. (1992). The Caenorhabditis elegans unc-93 gene encodes a putative transmembrane protein that regulates muscle contraction. J. Cell Biol. 117, 143–155. Abstract Article
Levin, J.Z., and Horvitz, H.R. (1993). Three new classes of mutations in the Caenorhabditis elegans muscle gene sup-9. Genetics 135, 53–70. Abstract
Levitan, D., and Greenwald, I. (1995). Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer's disease gene. Nature 377, 351–354. Abstract Article
Li, L., Linning, R.M., Kondo, K., and Honda, B.M. (1998). Differential expression of individual suppressor tRNA(Trp) gene family members in vitro and in vivo in the nematode Caenorhabditis elegans. Mol. Cell Biol. 18, 703–709.
Lissemore, J.L., Currie, P.D., Turk, C.M., and Maine, E.M. (1993). Intragenic dominant suppressors of glp-1, a gene essential for cell-signaling in Caenorhabditis elegans, support a role for cdc10/SW16/Ankyrin motifs in GLP-1 function. Genetics 135, 1023–1034. Abstract
Liu, J., Schrank, B., and Waterston, R.H. (1996). Interaction between a putative mechanosensory membrane channel and a collagen. Science 273, 361–364. Abstract
Lundquist, E.A., and Herman, R.K. (1994). The mec-8 gene of Caenorhabditis elegans affects muscle and sensory neuron function and interacts with three other genes: unc-52, smu-1 and smu-2. Genetics 138, 83–101. Abstract
Maine, E.M., and Kimble, J.E. (1989). Identification of genes that interact with glp-1, a gene required for inductive cell interactions in Caenorhabditis elegans. Development 106, 133–143. Abstract
Maine, E.M., and Kimble, J.E. (1993). Suppressors of glp-1, a gene required for cell communication during development in Caenorhabditis elegans, define a set of interacting genes. Genetics 135, 1011–1122. Abstract
Maruyama, I.N., Miller, D.M., and Brenner, S. (1989). Myosin heavy chain gene amplification as a suppressor mutation in Caenorhabditis elegans. Mol. Gen. Genet. 219, 113–118. Abstract Article
Moerman, D.G., Plurad, S., Waterston, R.H., and Baillie, D.L. (1982). Mutations in the unc-54 myosin heavy chain gene of Caenorhabditis elegans that alter contractility but not muscle structure. Cell 29, 773–781. Abstract Article
Nishiwaki, K., and Miwa, J. (1998). Mutations in genes encoding extracellular matrix proteins suppress the emb-5 gastrulation defect in Caenorhabditis elegans. Mol. Gen. Genet. 1259, 2–12. Abstract
Novelli, J., Ahmed, S., and Hodgkin, J. (2004). Gene interactions in C. elegans define DPY-31 as a candidate procollagen C-proteinase and SQT-3/ROL-4 as its predicted major target. Genetics 168, 1259–1273. Abstract Article
Park, E.C., and Horvitz, H.R. (1986). Mutations with dominant effects on the behavior and morphology of the nematode Caenorhabditis elegans. Genetics 113, 821–852. Abstract
Pilgrim, D.B., and Bell, J.B. (1993). Expression of a Drosophila melanogaster amber suppressor tRNA(Ser) in Caenorhabditis elegans. Mol. Gen. Genet. 241, 26–32. Abstract Article
Riddle, D.L., and Brenner, S. (1978). Indirect suppression in Caenorhabditis elegans. Genetics 89, 299–314. Abstract
Rogalski, T.M., Gilchrist, E.J., Mullen, G.P., and Moerman, D.G. (1995). Mutations in the unc-52 gene responsible for body wall muscle defects in adult Caenorhabditis elegans are located in alternatively spliced exons. Genetics 139, 159–169. Abstract
Roller, A.B., Hoffman, D.C., and Zahler, A.M. (2000). The allele-specific suppressor sup-39 alters use of cryptic splice sites in Caenorhabditis elegans. Genetics 154, 1169–1179. Abstract
Run, J.Q., Steven, R., Hung, M.S., van Weeghel, R., Culotti. J.G., and Way J.C. (1996). Suppressors of the unc-73 gene of Caenorhabditis elegans. Genetics 143, 225–236. Abstract
Selfors, L.M., Schutzman, J.L., Borland, C.Z., and Stern, M.J. (1998). soc-2 encodes a leucine-rich repeat protein implicated in fibroblast growth factor receptor signaling. Proc. Natl. Acad. Sci. USA 95, 6903–6908. Abstract Article
Spartz, A.K., Herman, R.K., and Shaw, J.E. (2004). SMU-2 and SMU-1, Caenorhabditis elegans homologs of mammalian spliceosome-associated proteins RED and fSAP57, work together to affect splice site choice. Mol. Cell Biol. 24, 6811–6823. Abstract Article
Sternberg, P.W., and Han, M. (1998). Genetics of RAS signaling in C. elegans. Trends Genet. 14, 466–472. Abstract Article
Sundaram, M., and Greenwald, I. (1993). Suppressors of a lin-12 hypomorph define genes that interact with both lin-12 and glp-1 in Caenorhabditis elegans. Genetics 135, 765–783. Abstract
Sundaram, M., and Han, M. (1995). The C. elegans ksr-1 gene encodes a novel Raf-related kinase involved in Ras-mediated signal transduction. Cell 83, 889–901. Abstract Article
Tax, F.E., Thomas, J.H., Ferguson, E.L., and Horvitz, H.R. (1997). Identification and characterization of genes that interact with lin-12 in Caenorhabditis elegans. Genetics 147, 1675–1695. Abstract
Treinin, M., Gillo, B., Liebman, L., and Chalfie, M. (1998). Two functionally dependent acetylcholine subunits are encoded in a single Caenorhabditis elegans operon. Proc. Natl. Acad. Sci. USA 95, 15492–15495. Abstract Article
Wang, X., Yang, C., Chai, J., Shi, Y., and Xue, D. (2002). Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science 298, 1587–1592. Abstract Article
Waterston, R.H. (1981). A second informational suppressor, sup-7 X, in Caenorhabditis elegans. Genetics 97, 307–325. Abstract
Waterston, R.H., and Brenner, S. (1978). A suppressor mutation in the nematode acting on specific alleles of many genes. Nature 275, 715–719. Abstract Article
Wen, C., Metzstein, M.M., and Greenwald, I. (1997). SUP-17, a Caenorhabditis elegans ADAM protein related to Drosophila KUZBANIAN, and its role in LIN-12/NOTCH signalling. Development 124, 4759–4767. Abstract
Wen, C., Levitan, D., Li, X., and Greenwald, I. (2000). spr-2, a suppressor of the egg-laying defect caused by loss of sel-12 presenilin in Caenorhabditis elegans, is a member of the SET protein subfamily. Proc. Natl. Acad. Sci. USA 97, 14524–14529. Abstract Article
Wills, N., Gesteland, R.F., Karn, J., Barnett, L., Bolten, S., and Waterston, R.H. (1983). The genes sup-7 X and sup-5 III of C. elegans suppress amber nonsense mutations via altered transfer RNA. Cell 33, 575–583. Abstract Article
Wu, Y., and Han, M. (1994). Suppression of activated Let-60 ras protein defines a role of Caenorhabditis elegans Sur-1 MAP kinase in vulval differentiation. Genes Dev. 8, 147–159. Abstract
Zahler, A.M., Tuttle, J.D., Chisholm, A.D. (2004, August). Genetic suppression of intronic +1G mutations by compensatory U1 snRNA changes in Caenorhabditis elegans. Genetics. 167(4), 1689–1696. Abstract Article
*Edited by Philip Anderson. Last revised March 29, 2005. Published December 27, 2005. This chapter should be cited as: Hodgkin, J. Genetic suppression (December 27, 2005), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.59.1, http://www.wormbook.org.
Copyright: © 2005 Jonathan Hodgkin. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: [email protected]
 All journal content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All journal content, except where otherwise noted, is licensed under a Creative Commons Attribution License.