Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergAbstract
The most abundant synapses in the central nervous system of vertebrates are inhibitory synapses that use the neurotransmitter γ-aminobutyric acid (GABA). GABA is also an important neurotransmitter in C. elegans; however, in contrast to vertebrates where GABA acts at synapses of the central nervous system, in nematodes GABA acts primarily at neuromuscular synapses. Specifically, GABA acts to relax the body muscles during locomotion and foraging and to contract the enteric muscles during defecation. The importance of this neurotransmitter for basic motor functions of the worm has facilitated the genetic analysis of proteins required for GABA function. Genetic screens have identified the GABA biosynthetic enzyme, the vesicular transporter, inhibitory and excitatory receptors, and a transcription factor required for the differentiation of GABA cell identity. The plasma membrane transporter and other GABA receptors have been identified by molecular criteria.
γ-aminobutyric acid (GABA) is an amino acid neurotransmitter synthesized by decarboxylation of glutamate by the enzyme glutamic acid decarboxylase (Figure 1). GABA had been long known to exist in plants and bacteria, where it serves a metabolic role in the Krebs cycle. In 1950, Eugene Roberts and Jorge Awapara independently discovered that there were prodigious amounts of GABA in the mammalian central nervous system — 1 mg per gram — and GABA was virtually undetectable in other tissues (for historical perspectives, see Roberts, 2000 and Florey, 1991). However, GABA was not accepted as neurotransmitter until the 1960's after a great deal of physiological experimentation. In 1953, Ernst Florey observed that an unknown compound from horse brain inhibited the crayfish stretch receptor when applied exogenously (Florey, 1953). Florey demonstrated that this same fraction inhibited the patellar reflex in cats and thus extended the action of this factor to vertebrates (Florey and McLennan, 1955). With the help of Alva Bazemore from Merck Inc. Florey purified the molecule from 100 pounds of cow brain and determined that the active compound was GABA. They further demonstrated that chemically synthesized GABA could inhibit the crayfish stretch receptor (Bazemore et al., 1957). This observation prompted Ernst Florey to propose that GABA was acting as an inhibitory neurotransmitter in the brain. However, GABA's extraordinary qualities soon became a burden to its acceptance into the pantheon of neurotransmitters; its enormous abundance in the vertebrate brain — 1000-fold higher than known monoamine neurotransmitters, its simple structure and and its role in the Krebs cycle (the "GABA shunt") suggested that it was likely to be involved in metabolism rather than signaling. Moreover, it was demonstrated that GABA was not responsible for blocking the cat knee-jerk response (which is mediated by glycine that contaminated Florey's early preparations; Curtis et al., 1959). Finally, GABA was not found in significant concentrations in any invertebrates. Thus, GABA did not act as a neurotransmitter in organisms that made it; and it was not found in organisms in which it acted. GABA no longer fulfilled the qualifications of a neurotransmitter and by 1960 it had been demoted to a mere metabolite (Edwards et al., 1999a).
|
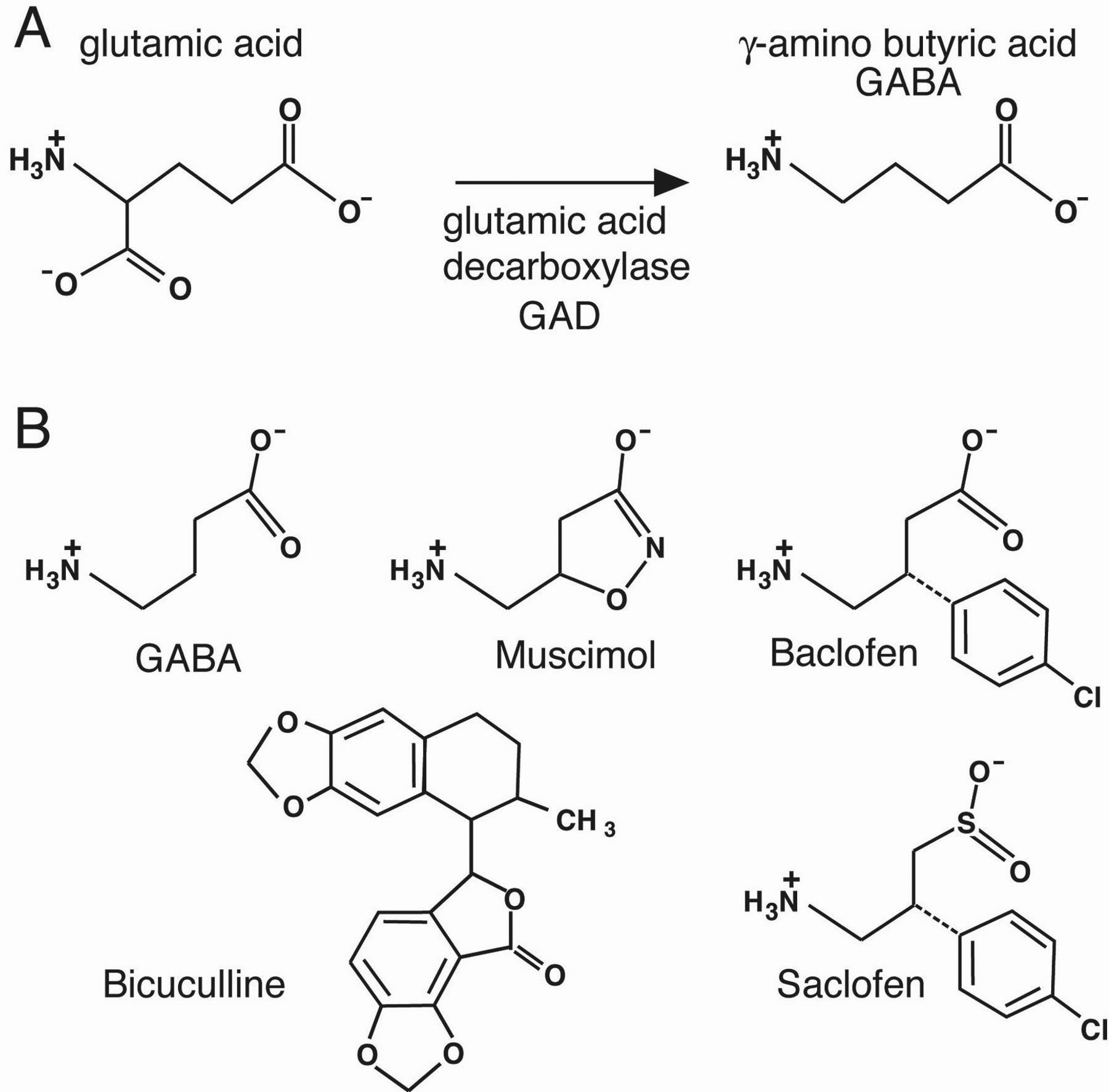
Figure 1. GABA structure. (A) GABA is synthesized from glutamate by the enzyme glutamic acid decarboxylase (GAD). (B) GABA drugs. Muscimol is a GABAA agonist and baclofen is a GABAB agonist. Bicuculline and saclofen are antagonists of GABAA and GABABrespectively. Invertebrate GABAA-like receptors are insensitive to bicuculline.
Only a few years later, in part due to studies in nematodes, GABA was rehabilitated as a potential neurotransmitter. First, del Castillo and colleagues, working with the parasitic nematode Ascaris, demonstrated that application of GABA [10 uM] or the GABA mimic piperazine [1mM] potently abolished spontaneous action potentials in muscles (Del Castillo et al., 1964a; Del Castillo et al., 1964b; Del Castillo et al., 1963). This inhibition was caused by a hyperpolarization of the muscle from a resting potential of -30mV to -45mV, mediated by increased chloride conductance. Furthermore, stimulation of the inhibitory motor neurons in Ascaris and crayfish duplicated the effect observed by GABA application (Del Castillo et al., 1967; Otsuka et al., 1966). GABA's presumed absence in invertebrates was contradicted by the discovery that it was found to be highly concentrated in inhibitory motor neurons of Ascaris (Guastella et al., 1991; Johnson and Stretton, 1987) and crayfish (Kravitz et al., 1963). Furthermore, lysates from C. elegans exhibited enzymatic activity from the GABA biosynthetic enzyme, glutamic acid decarboxylase (Hedgecock, 1976).
Electrophysiological recordings from Ascaris by del Castillo demonstrated that GABA application inhibits body muscles by opening chloride channels (Del Castillo et al., 1963). It is now known that these actions of GABA are mediated by the GABAA receptor, a GABA-gated chloride channel (Schofield et al., 1987). In vertebrate neurons, chloride ions are pumped out of the cell; thus, activation of GABA receptors will permit chloride to diffuse into the cell, hyperpolarize the membrane and decrease the excitability of the cell. This type of inhibition is called hyperpolarizing inhibition. In some cells, internal chloride is at concentrations higher than the equilibrium potential. In these cases, opening chloride channels caused an efflux of this anion, creating an inward current, and depolarizing the membrane. Nevertheless, this depolarization still inhibits muscle contraction because the increase in chloride conductance creates a 'current shunt' for excitatory currents (Kuffler and Eyzaguirre, 1955). Specifically, the abundance of open chloride channels clamps the membrane voltage at the chloride equilibrium potential and 'shunts' further depolarization. In addition, the weak depolarization caused by chloride conductance inactivates Na+ channels (Edwards et al., 1999b). This type of inhibition is called depolarizing inhibition. Thus, activation of GABA receptors will inhibit cell activity whether it hyperpolarizes or depolarizes the cell.
GABA can also inhibit membrane excitability by opening K+ channels and inhibiting Ca++ channels. These actions are mediated by the GABAB receptor, a 7-pass transmembrane receptor (Kaupmann et al., 1997; for a review see Calver et al., 2002). A functional GABAB receptor is composed of two subunits that form a heterodimer. GABA binding to the GABAB receptor activates the trimeric G protein Gi/o (Campbell et al., 1993). The activated Gαo subunit can inhibit voltage-gated Ca++ channels or activate K+ channels.
The actions of the different GABA receptor types were originally defined by independent pharmacological sensitivities. GABAA receptors are activated by muscimol, whereas GABAB receptors are activated by baclofen (Figure 1). The GABAA receptors are blocked by bicuculline, whereas the GABAB receptor can be blocked by saclofen or phaclofen (Guillon et al., 1999). Although the pharmacology of GABA receptors in C. elegans differs somewhat compared to GABA receptors in vertebrates, nematodes possess both GABAA and GABAB receptors based on sequence similarity.
To identify the GABA-containing cells in C. elegans, wild-type animals were stained with antibodies against the neurotransmitter. Antibody staining revealed that 26 of the 302 neurons present in C. elegans express the neurotransmitter GABA (Figure 2A). These 26 GABA neurons are comprised of 6 DD, 13 VD, 4 RME, RIS, AVL and DVB (Figure 2B). These neurons fall into different classes based on their synaptic outputs: the D-type neurons, that is, the 6 DD and 13 VD motor neurons, innervate the dorsal and ventral body muscles, respectively; the 4 RME motor neurons innervate the head muscles; the AVL and DVB motor neurons innervate the enteric muscles; and RIS is an interneuron (White et al., 1986).
Although Ascaris and C. elegans are diverged by 500 million years, their nervous systems are remarkably conserved. In Ascaris, a nearly equivalent set of 26 major GABA-synthesizing neurons were identified by immunostaining (Guastella et al., 1991; Johnson and Stretton, 1987). In the ventral nerve cord 6 DD-like and 13 VD-like cells express GABA. In the head, there are 4 RME-like cells and in the tail a DVB-like cell. Variable staining was observed for additional cells in the head; although there are 6 bilaterally symmetric cells which stained in the cephalic ganglia, they do not appear to be homologs of AVL and RIS.
|
Figure 2. The GABA nervous system. (A) Fluorescent micrograph of an adult C. elegans hermaphrodite stained with an antiGABA antiserum. There are 26 neurons that stain for GABA. Anterior is to the right and the right side of the body is shown. Scale bar = 0.10 mm. (B) Schematic drawing of the positions of the 26 GABA-containing neurons. The DD and VD motor neurons innervate the body muscles, the RME neurons innervate the head muscles, AVL and DVB innervate the enteric muscles, and RIS is an interneuron. Reprinted from Schuske et al. (2004), Copyright (2004), with permission from Elsevier.
To determine the in vivo behavioral role of the GABA neurons, laser ablation of specific classes of neurons was performed (McIntire et al., 1993). Functions were identified for the D-type body muscle motor neurons, for the RME head muscle motor neurons, and for the AVL and DVB enteric muscle motor neurons. No function was identified for the RIS interneuron. Based on these studies, it was concluded that GABA acts as both an inhibitory and excitatory neurotransmitter in C. elegans.
The 19 ventral cord D-type neurons inhibit contraction of the ventral and dorsal body wall muscles during locomotion (McIntire et al., 1993). A bend in the body is made by contracting muscles on one side of the body while relaxing muscles via GABA innervation on the opposite side (Figure 3C), which gives the worm its distinctive sinusoidal body posture. Movement occurs when a body bend is propagated from one end of the animal to the other (Figure 3A). Because the VD and DD neurons are required for relaxation of the body wall muscles, when these neurons are killed, the operated worm only has excitatory input into its muscles. Operated animals can still adopt a sinusoidal posture and can swim, albeit with reduced wave amplitudes. However, when an animal lacking D-type neuron function is touched, it "shrinks", that is, it pulls in its head and shortens its body due to hypercontraction of the body wall muscles on both sides of the body (Figure 3B). Therefore, GABA released from the ventral cord D-type neurons is required to relax muscles, to reset posture when changing directions, and provide normal wave shape when swimming.
|
Figure 3. The D-type motor neurons inhibit body muscles. (A) When a wild-type worm is tapped on the nose it moves backwards by propagating a sinusoidal wave from the tail to the head. (B) When a mutant defective for GABA transmission is tapped on the nose, it contracts muscles on both sides of its body causing the animal to shrink. Scale 0.5 mm. (Photo courtesy of J. White). (C) Motor neuron connectivity. Motor neuron cell bodies are in the ventral cord. The DD and VD GABA neurons (blue) synapse to the dorsal and ventral body muscles, respectively. Cholinergic motor neurons (red) send inputs to the ventral and dorsal body muscles, as well as the GABA motor neurons. Release of acetylcholine leads to contraction of the body wall muscle on one side and stimulates GABA release onto muscles on the opposite side. This stimulation and contralateral inhibition causes the body to bend and leads to coordinated locomotion. Excitatory inputs are indicated by dark shading. Reprinted from Schuske et al. (2004), Copyright (2004), with permission from Elsevier.
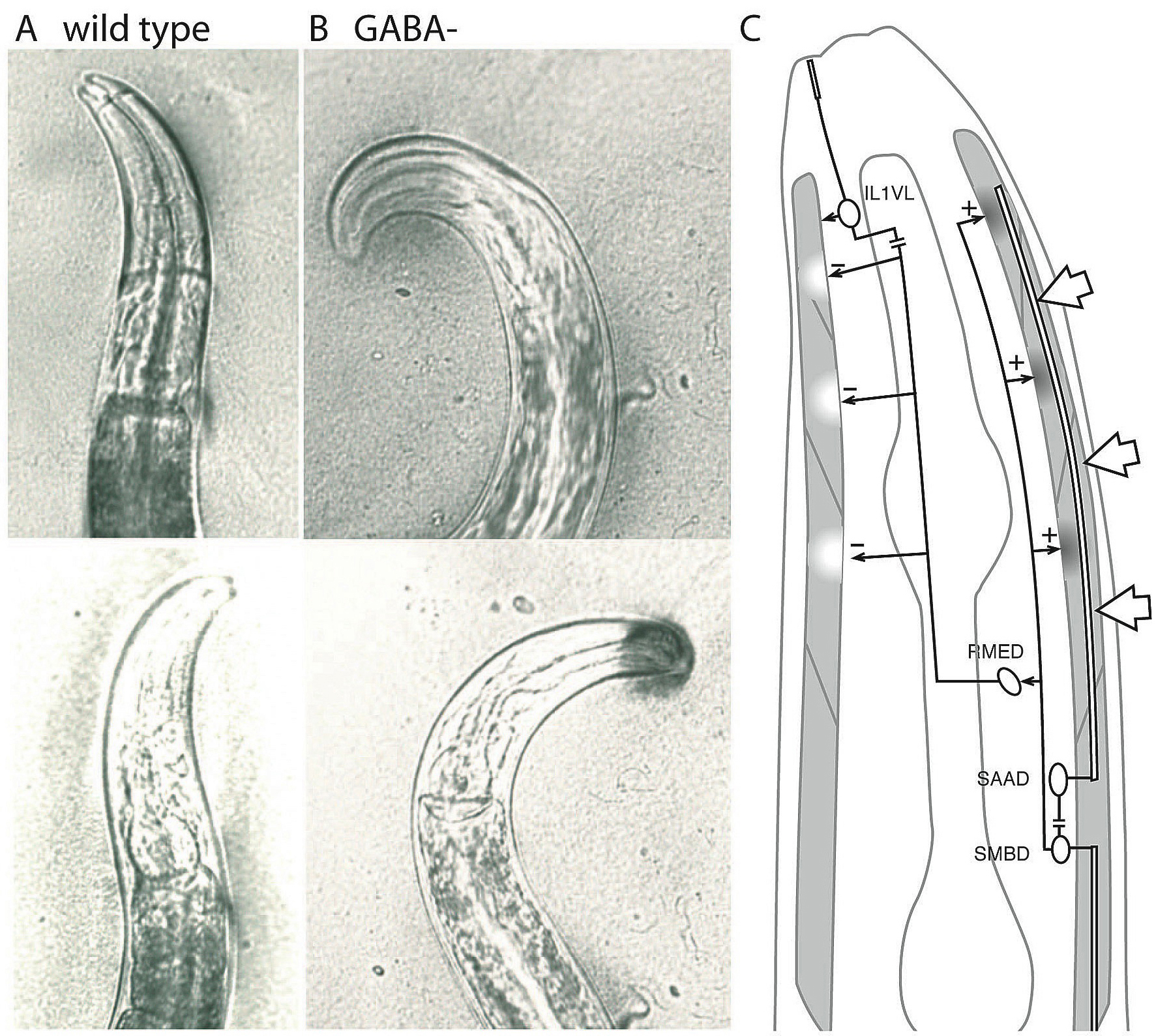
The RME neurons regulate foraging movements of the head. During normal foraging the tip of the nose moves from side to side within a narrow arc of movement (Figure 4A). Killing the four RME motor neurons by laser microsurgery resulted in worms with "loopy" foraging in which the flexures of the nose became grossly exaggerated (Figure 4B). Thus, the RMEs appear to limit the extent of head deflection during foraging. The RMEs receive input from sensory neurons of the head and nose; for example, SMB and SAA innervate the ipsilateral RME, either directly or indirectly (Figure 4C). Both of these neurons have long undifferentiated processes that run anteriorly and posteriorly along the dorsal side of the head, and these neurons may provide proprioceptive information about head position (White et al., 1986). The RMEs send outputs to contralateral muscles of the head. It is possible that bending of the head during foraging activates stretch receptors in the SMB and SAA neurons, which then activate an RME neuron, which in turn relaxes the muscles on the opposite side of the bend and restores head posture.
|
Figure 4. The RME motor neurons inhibit foraging movements. (A) Wild-type worms wave their heads in a narrow arc while foraging. (B) Mutants that lack GABA curve their heads in exaggerated flexures when foraging. (C) RMED motor neuron connectivity (White et al., 1986). The long undifferentiated processes of SAA and SMB may contain stretch receptors that are activated by positive curvature. These could stimulate the RME neuron which releases GABA onto the contralateral side to relax the flexed muscles. The RMED and RMEV neurons also have processes extending into the dorsal and ventral nerve cords, respectively. In addition, the dorsal and ventral RMEs receive input from the mechanosensory neurons IL1 and IL2, and the lateral RMEs receive input from the mechanosensory neurons OLQ and OLL, which could cause the head to move away from a stimulus by causing the muscles to relax on the touched side. Open arrows, stretch input; horizontal lines, gap junctions; solid arrows, chemical synapses. Excitatory inputs are indicated by dark shading in the muscle, inhibitory inputs are shown as white areas.
The GABA motor neurons, AVL and DVB, stimulate the enteric muscles during defecation (Figure 5B; McIntire et al., 1993). Every 50 seconds a worm undergoes a stereotypic pattern of muscle contractions, which ultimately leads to the expulsion of intestinal contents (Figure 5A; Thomas, 1990). The final step of the motor program is the contraction of the enteric muscles (Emc). This contraction squeezes the intestine and opens the anus so that the intestinal contents can be expelled. AVL and DVB are required in the last step to contract the enteric muscles (McIntire et al., 1993). When these neurons are killed using a laser, the worm fails to contract its enteric muscles and becomes bloated with food. Addition of GABA or the GABA agonist muscimol will restore contractions of this muscle. Therefore, GABA released from AVL and DVB is required to induce contraction of these muscles.
|
Figure 5. AVL and DVB motor neurons stimulate enteric muscles. (A) Schematic diagram of the defecation motor program (Thomas, 1990). Every 50s an animal initiates a defecation cycle, which consists of the three stereotyped independent muscle contractions. The cycle begins with the posterior body contraction (pBoc), followed by the anterior body contraction (aBoc), and concluded by the enteric muscle contraction (Emc). Contracting muscles are indicated by dark shading. (B) Schematic of the enteric muscles. The enteric muscles consist of four cells: the anal depressor, sphincter, and two bilaterally-symmetric intestinal cells. These cells are gap-junctioned to one another, and each cell sends a muscle arm to the preanal ganglion where DVB makes synaptic contact. AVL, which is located anteriorly in the ventral head ganglion, sends a process along the ventral nerve cord abutting DVB and the enteric muscles. Release of GABA from AVL and DVB causes the anal depressor and intestinal muscles to contract. Reprinted from Schuske et al. (2004), Copyright (2004), with permission from Elsevier.
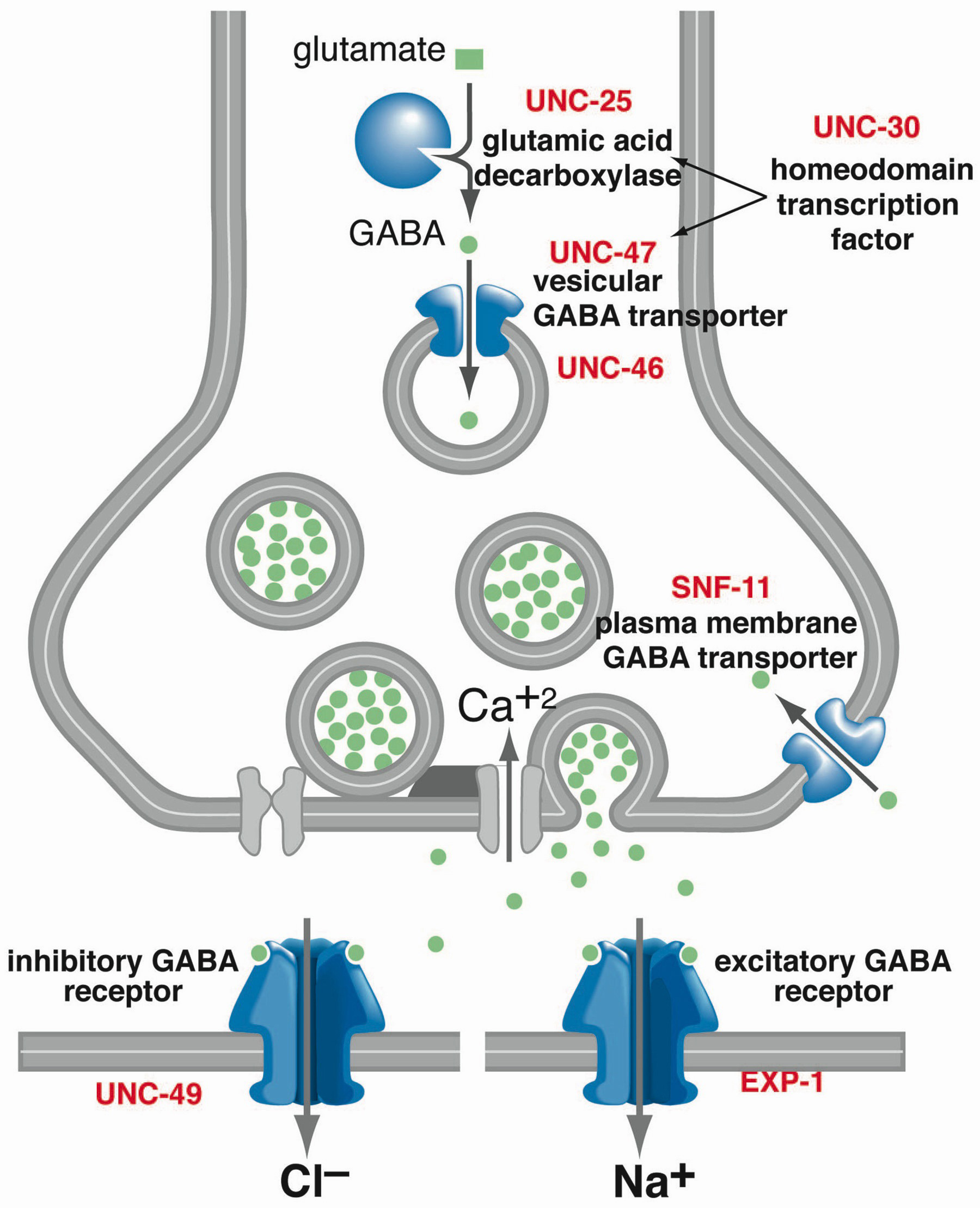
To identify genes required for GABA function, mutants were identified that resembled worms in which the GABA neurons were killed (McIntire et al., 1993; Thomas, 1990). A total of six genes were identified that caused all or a subset of the behavioral defects when mutated (Table 1). Three of these mutants (unc-25, unc-46 and unc-47) were shrinkers and expulsion defective, suggesting that these genes were important for universal functions of GABA. Two of these mutants (unc-30 and unc-49) were shrinkers but had normal expulsions indicating that they specifically eliminated the inhibitory functions. One of these mutants (exp-1) was only defective for the enteric muscle contractions indicating that it was specific for the excitatory functions of GABA (Thomas, 1990). Shrinking or expulsion-defective mutants that also exhibited pleiotropic phenotypes were ignored.
Table 1. GABA mutant phenotypes
| Mutant | Inhibitory function presenta | Excitatory function present[a] | Muscimol sensitive[a] | GABA levels[a] | Protein | Reference | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| unc-25 | - | - | yes | (-) | GAD | Jin, 1999 | ||||
| unc-30 | - | + | yes |
|
Transcription factor | Jin, 1994 | ||||
| unc-46 | - | - | yes | (+) | ? | N/A | ||||
| unc-47 | - | - | yes | (++++) | VGAT | McIntire, 1997 | ||||
| unc-49 | - | + | no | (+) | Anionic GABA-R | Bamber, 1999 | ||||
| exp-1 | + | - | no | (+) | Cationic GABA-R | Beg, 2003 | ||||
|
a- = defective behavior, + = wild type behavior, (+) = wild type GABA level, (-) = low GABA level, (++++) = high GABA level (McIntire et al., 1993). |
||||||||||
unc-25 encodes the biosynthetic enzyme for GABA, glutamic acid decarboxylase (GAD; Figure 6; Jin et al., 1999). Previously, in vitro data had indicated that GABA could be involved in neuronal development, specifically in cell proliferation, cell migration, neurite extension, or synapse formation (Belhage et al., 1998). However, genetic studies in C. elegans (below) and more recently in mice have demonstrated that GABA does not play a role in development (Ji et al., 1999).
Careful analysis of the unc-25 mutant phenotype failed to reveal defects in neuronal development. Axon morphology of GABA neurons is normal in adult unc-25 mutants (Jin et al., 1999). Synaptic varicosities, assayed by clusters of fluorescently-tagged synaptic vesicles, are at normal positions and densities in the ventral cord GABA motor neurons (Jin et al., 1999). The post-synaptic GABA receptor also has a normal distribution in unc-25 mutants (Gally and Bessereau, 2003). A reconstruction of the ventral nerve cord from serial electron micrographs demonstrated that the synaptic ultrastructure and connectivity were correct (Jin et al., 1999). Definitive proof that GABA synapses are functional was provided by rescuing unc-25 function as an adult: addition of exogenous GABA provided the AVL and DVB neurons with an external source of GABA, GABA was pumped into cells via the plasma membrane transporter, and normal behavior was restored (McIntire et al., 1993). These data demonstrate unequivocally that GABA release is not required for axon outgrowth, synapse formation or synapse maintenance in C. elegans.
The unc-47 gene encodes the transporter that pumps GABA into the synaptic vesicle (Figure 6; McIntire et al., 1997) UNC-47 is an 11-pass transmembrane protein which was not related to other transporter families known at the time. The rat homolog of UNC-47 was cloned by homology and it was found that rUNC-47 (currently called VIAAT or VGAT) is capable of transporting GABA and glycine into acidified vesicles (McIntire et al., 1997; Sagne et al., 1997). The discovery of UNC-47 made possible the identification several families of amino acid transporters based on homology to UNC-47. The System N and A transporters (SLC38 family of solute carrier proteins) transport neutral amino acids across the plasma membrane using the Na+ gradient for coupled transport (N: glutamine, asparagine, and histidine; A: alanine asparagine cysteine, glutamine, glycine, methionine, and serine; Mackenzie and Erickson, 2004). The SLC36 family transports amino acids from the lysosome or at the apical membrane of the intestine using the H+ ion gradient for coupled transport (Boll et al., 2004). Like all other vesicular neurotransmitter transporters, UNC-47 uses a proton gradient generated by the vacuolar ATPase as a secondary energy source to transport GABA against its concentration gradient. Surprisingly, there are two other unrelated families of proteins which transport neurotransmitters into synaptic vesicles: the vesicular monoamine and acetylcholine transporter family (Duerr et al., 1999; Liu et al., 1992), and the glutamate transporter family (Lee et al., 1999; Takamori et al., 2000). Why these classes of neurotransmitter transporters, which all use a proton gradient for an energy source, did not evolve from a common ancestor is a mystery. Moreover, these transporters are also unrelated to the plasma membrane neurotransmitter transporters. Plasma membrane neurotransmitter transporters remove secreted neurotransmitters from the synaptic cleft by pumping them back across the plasma membrane, but use the Na+ gradient for active transport instead of a H+ gradient (see 'Other GABA genes' below).
|
Figure 6. Genes required for GABA function in C. elegans. GABA is synthesized in the cytoplasm of the neuron by glutamic acid decarboxylase (GAD) encoded by the unc-25 gene. GABA is then transported into the synaptic vesicle by the vesicular GABA transporter (VGAT) encoded by the unc-47 gene. The UNC-46 protein is likely to be an accessory protein in vesicular GABA transport. The UNC-30 transcription factor is required for GAD and VGAT expression in the GABA motor neurons. GABA released from the DD and VD motor neurons activates the inhibitory GABAA-like UNC-49 receptor; the influx of chloride ions causes the relaxation of the body muscles. GABA release from the AVL and DVB motor neurons activates the novel excitatory EXP-1 receptor; the influx of sodium ions causes the contraction of the enteric muscles GABA is cleared from the cleft by the plasma membrane transporter, SNF-11. Adapted from Schuske et al. (2004), Copyright (2004), with permission from Elsevier.
unc-46 mutants are defective for both the inhibitory and excitatory functions of GABA which suggests that UNC-46 acts presynaptically (McIntire et al., 1993). However, the phenotype of unc-46 mutants is weaker than unc-25 and unc-47 mutants. Overexpression of the UNC-47 GABA transporter in an unc-46 mutant background partially rescues the defects in these animals (K. Schuske and E. Jorgensen, unpublished). Thus, UNC-46 may regulate the activity or localization of the vesicular GABA transporter but it is not absolutely required for transport activity (Figure 6).
The biosynthetic enzyme and vesicular transporter for GABA are the key enzymes that determine neurotransmitter-specific identity for GABA neurons. The unc-30 gene is a transcription factor that coordinately regulates the expression of these enzymes. UNC-30 was the founding member of a class of homeodomain-containing transcription factors that include Pitx1, Pitx2 and Pitx3 in vertebrates (Jin et al., 1994). The unc-30 gene is expressed in the VD and DD neurons and these cells show defects in GABA cell identity in unc-30 mutants. First, axonal outgrowth of the D neurons is defective in unc-30 mutants (McIntire et al., 1993). Second, selection of synaptic targets is abnormal in these mutants (J. White, personal communication). Third, neither GAD, the biosynthetic enzyme for GABA, nor the vesicular GABA transporter are expressed in the VD and DD neurons in unc-30 mutants (Eastman et al., 1999). The UNC-30 protein binds to promoter elements in both the unc-25/ GAD and unc-47 /VGAT genes, and these elements were necessary for expression of these genes in the DD and VD neurons (Eastman et al., 1999). Ubiquitous expression of the UNC-30 protein led to the ectopic synthesis of GABA in many non-GABA neurons as well as in some non-neuronal tissues. These data suggest that UNC-30 is sufficient to express GABA neuron identity in cells, even if these cells, such as hypodermal cells, lack factors for neuronal cell identity (Jin et al., 1994). It is thus surprising that UNC-30 is expressed in six cells that do not express GABA cell identity - why don't these neurons express unc-25 or unc-47 given that UNC-30 expression is sufficient for activation of these genes in epidermal cells? The other GABA neurons, AVL, DVB, RIS and the RMEs, do not require UNC-30 for the expression of unc-25 and unc-47. In lim-6 mutants, unc-25 expression is reduced in DVB and RIS, but unc-47 expression is normal; moreover, lim-6 is involved in the fate of nonGABA and non-neuronal cells as well, suggesting it does not specifiy neurotransmitter type (Hobert et al., 1999). GABA neuron identity in these cells requires other as yet unidentified genes. Other genes specify cell identities among GABA neurons; for example, ahr-1 specifies RMEL/R fates as different from an RMED/V ground state (Huang et al., 2004); unc-55 specifies VD cell identity as unique from a DD ground state (Zhou et al., 1998; Shan et al.; 2005). However, once again, these transcription factors do not define the GABA neurotransmitter identity in these cells.
What are all the targets of UNC-30 in the genome? Microarray expression studies of GABA neurons combined with computational searches for UNC-30 binding motifs revealed 24 potential UNC-30 target genes, which were not identified in the GABA screen (Cinar et al., 2005). Although many of these are not unique to GABA neurons, analysis of expression patterns indicate that at least some of these require UNC-30 for expression in GABA neurons.
In summary, UNC-30 is required for GABA neuron specification in the D-type neurons and its expression is sufficient for conferring GABA neuron identity. However, its role in cell identity is complicated. UNC-30 is not required by all GABA neurons for GABA cell identity. How these cells regulate neurotransmitter specificity is not known. Moreover, some cells that express UNC-30 do not display GABA cell identity. Why these cells do not express GABA specific genes is not known.
GABAA receptors are GABA-gated chloride channels that inhibit cell activity. The GABAA receptor, that inhibits body muscle contraction during locomotion, is encoded by the unc-49 gene (Figure 6; Bamber et al., 1999; Bamber et al., 2005). The unc-49 locus encodes three distinct GABA receptor subunits by splicing a common N-terminal ligand-binding domain to one of three alternative C-terminal domains, producing the UNC-49A, UNC-49B, and UNC-49C subunits (Bamber et al., 1999). Keep in mind that these alternative gene products are all subunits of a GABAA ligand-gated ion channel and are not related to GABAB receptors. This unusual gene structure is conserved in the distantly-related nematode C. briggsae. The UNC-49B and UNC-49C subunits are expressed in the muscles and localized to synapses from the D-type GABA motor neurons (Bamber et al., 1999; Bamber et al., 2005; Gally and Bessereau, 2003). The GABA receptor at neuromuscular junctions is a heteromer composed of the B and C subunits (Bamber et al., 2005). The B subunit is required for localization of the receptor to neuromuscular junctions and the C subunit imparts specific pharmacological properties to the heteromeric receptor (Bamber et al., 2005; Bamber et al., 2003). The UNC-49A subunit is barely detectable in vivo, and does not heteromultimerize with UNC-49B or UNC-49C to form a functional receptor in vitro (Bamber et al., 1999).
Mutations in exp-1 were originally identified in screens for mutants with defects in the defecation cycle (Thomas, 1990). exp-1 mutants exhibit defects in excitatory GABA functions but are normal for the inhibitory GABA functions. Specifically, exp-1 mutants lack enteric muscle contractions yet move and forage normally, and thus resemble animals in which the AVL and DVB GABA motor neurons were killed (McIntire et al., 1993). Consistent with its role in excitatory GABA transmission, it was found that exp-1 encodes a novel GABA-gated cation channel (Figure 6; Beg and Jorgensen, 2003). The EXP-1 protein is expressed in the enteric muscles, and is localized to the synaptic inputs from the GABA neurons AVL and DVB. Electrophysiological characterization of the EXP-1 GABA receptor demonstrated that the channel is selective for cations (Beg and Jorgensen, 2003), in contrast to all the other GABA-gated ion channels, which are anion selective (Olsen, 1991). The EXP-1 protein resembles conventional GABA receptors except in the pore-forming domain which contains amino acid changes which permit cation permeability (Keramidas et al., 2000, 2002).
Examination of the genome indicates that there are several genes involved in GABA function that were not identified in the genetic screens for GABA function. There are multiple GABA receptor subunits that can be observed in the genome by sequence homology that were not mutated in screens for shrinker or expulsion-defective mutants. Notably, there are α and β GABAA receptor subunits (T19B10.10, ZC482.1), another UNC-49 like receptor subunit (F11H8.2), another EXP-1-related receptor subunit (Y46G5A.26, A. Beg and E. Jorgensen, unpublished), and there is a heteromeric metabotropic GABAB receptor encoded in the genome (Y41G9A.4, ZK180.1; J. Dittman and J. Kaplan, personal communication). The GABA-mediated behaviors controlled by these four additional GABA receptors are unknown; however, one of these receptors might be responsible for mediating foraging behavior, which is defective in unc-25/GAD mutants but is not defective in either unc-49 mutants or in exp-1 mutants.
Once neurotransmitters are secreted into the cleft, they must be cleared by active processes. GABA is removed from the cleft by a plasma membrane transporter which pumps the transmitter back into the neuron (Guastella et al., 1990). The plasma membrane GABA transporter was not identified in the genetic screens for shrinkers or for expulsion-defective mutants. Nevertheless, the activity of the GABA transporter could be observed. Specifically, the addition of exogenous GABA to mutants that lacked GABA (unc-25), restored GABA immunoreactivity and function (McIntire et al., 1993). Moreover, pharmacological antagonists of the GABA transporter blocked GABA uptake into neurons of unc-25. The gene snf-11 is highly similar to the vertebrate GABA transporter (Figure 6), and the transporter is expressed in all GABA neurons (Jiang et al., 2005). Expression of this transporter in heterologous cells confirmed that it is a functional plasma membrane GABA transporter (Jiang et al., 2005).
Nematodes and vertebrates diverged over 800 million years ago. Nevertheless the proteins governing GABA cell identity, biosynthesis and transport are conserved in the nematode and vertebrate nervous systems. Notably, studies in the nematode identified the vesicular GABA transporter and the UNC-30 homeodomain transcription factor, and subsequent genome comparisons identified the vertebrate orthologs of these genes. Although there does not appear to be a GABA-gated cation channel related to EXP-1 in the vertebrate genome, the GABAA chloride channel and the GABAB G-protein coupled receptor are both found in the vertebrate and nematode genomes.
Bamber, B.A., Beg, A.A., Twyman, R.E., and Jorgensen, E.M. (1999). The Caenorhabditis elegans unc-49 locus encodes multiple subunits of a heteromultimeric GABA receptor. J. Neurosci. 19, 5348–5359. Abstract
Bamber, B.A., Richmond, J.E., Otto, J.F., and Jorgensen, E.M. (2005). The composition of the GABA receptor at the Caenorhabditis elegans neuromuscular junction. Br. J. Pharmacol. 144, 502–509. Abstract Article
Bamber, B.A., Twyman, R.E., and Jorgensen, E.M. (2003). Pharmacological characterization of the homomeric and heteromeric UNC-49 GABA receptors in C. elegans. Br. J. Pharmacol. 138, 883–893. Abstract Article
Bazemore, A.W., Elliott, K.A. C., and Florey, E. (1957). Isolation of factor I. J. Neurochem. 1, 334–339.
Beg, A.A., and Jorgensen, E.M. (2003). EXP-1 is an excitatory GABA-gated cation channel. Nat. Neurosci. 6, 1145–1152. Abstract Article
Belhage, B., Hansen, G.H., Elster, L., and Schousboe, A. (1998). Effects of Gamma-aminobutyric acid (GABA) on synaptogenesis and synaptic function. Perspect Dev. Neurobiol 5, 235–246. Abstract
Boll, M., Daniel, H., and Gasnier, B. (2004). The SLC36 family: proton-coupled transporters for the absorption of selected amino acids from extracellular and intracellular proteolysis. Pflugers Arch. 447, 776–779. Abstract Article
Calver, A.R., Davies, C.H., and Pangalos, M. (2002). GABA(B) receptors: from monogamy to promiscuity. Neurosignals 11, 299–314. Abstract Article
Campbell, V., Berrow, N., and Dolphin, A.C. (1993). GABAB receptor modulation of Ca2+ currents in rat sensory neurones by the G protein G(0): antisense oligonucleotide studies. J. Physiol. 470, 1–11. Abstract
Cinar, H., Keles, S., and Jin, Y. (2005). Expression profiling of GABAergic motor neurons in Caenorhabditis elegans. Curr. Biol. 15, 340–346. Abstract Article
Curtis, D.R., Phillis, J.W., and Watkins, J.C. (1959). The depression of spinal neurones by Gamma-amino-η-butyric acid and b-alanine. J. Physiol. (Lond.) 146, 185–203.
Del Castillo, J., De Mello, W.C., and Morales, T. (1964a). Influence of some ions on the membrane potential of Ascaris muscle. J. Gen. Physiol. 48, 129–140. Article
Del Castillo, J., De Mello, W.C., and Morales, T. (1964b). Inhibitory action of Gamma-aminobutyric acid (GABA) on Ascaris muscle. Experientia 20, 141–143. Article
Del Castillo, J., De Mello, W.C., and Morales, T. (1967). The initiation of action potentials in the somatic musculature of Ascaris lumbricoides. J. Exp. Biol. 46, 263–279.
Del Castillo, J., Morales, T., and Sanchez, V. (1963). Action of piperazine on the neuromuscular system of Ascaris lumbricoides. Nature 200, 706–707.
Duerr, J.S., Frisby, D.L., Gaskin, J., Duke, A., Asermely, K., Huddleston, D., Eiden, L.E., and Rand, J.B. (1999). The cat-1 gene of Caenorhabditis elegans encodes a vesicular monoamine transporter required for specific monoamine-dependent behaviors. J. Neurosci. 19, 72–84.
Eastman, C., Horvitz, H.R., and Jin, Y. (1999). Coordinated transcriptional regulation of the unc-25 glutamic acid decarboxylase and the unc-47 GABA vesicular transporter by the Caenorhabditis elegans UNC-30 homeodomain protein. J. Neurosci. 19, 6225–6234. Abstract
Edwards, D.H., Heitler, W.J., and Krasne, F.B. (1999a). Crustacean studies and the early history of GABA. Trends Neurosci. 22, 347. Abstract Article
Edwards, D.H., Heitler, W.J., and Krasne, F.B. (1999b). Fifty years of a command neuron: the neurobiology of escape behavior in the crayfish. Trends Neurosci. 22, 153–161. Abstract Article
Florey, E. (1953). Ubereinen nervosen Hemmungsfaktor in Gehirn und Ruckenmark. Naturwissenschaften 40, 295–296. Article
Florey, E. (1991). GABA: history and perspectives. Can. J. Physiol. Pharmacol. 69, 1049–1056. Abstract
Florey, E., and McLennan, H. (1955). Effects of an inhibitory factor (Factor I) on central synaptic transmission. J. Physiol. 130, 446–455.
Gally, C., and Bessereau, J.L. (2003). GABA is dispensable for the formation of junctional GABA receptor clusters in Caenorhabditis elegans. J. Neurosci. 23, 2591–2599. Abstract
Guastella, J., Johnson, C.D., and Stretton, A.O. W. (1991). GABA-immunoreactive neurons in the nematode Ascaris. J. Comp. Neurol. 584–597. Abstract
Guastella, J., Nelson, N., Nelson, H., Czyzyk, L., Keynan, S., Miedel, M.C., Davidson, N., Lester, H.A., and Kanner, B.I. (1990). Cloning and expression of a rat brain GABA transporter. Science 249, 1303–1306. Abstract
Guillon, J., Levasseur, V., Sonnet, P., Stiebing, S. Boulouard, M., Dallemagne, P., Quermonne, M.-A., and Rault, S. (1999). Pharmacological Evaluation of New Baclofen Derivatives. Pharm. Pharmacol. Commun. 5, 243–247.
Hedgecock, E. (1976). GABA metabolism in Caenorhabditis elegans., Ph.D. Thesis, University of California, Santa Cruz.
Hobert, O., Tessmar, K., and Ruvkun, G. (1999). The Caenorhabditis elegans lim-6 LIM homeobox gene regulates neurite outgrowth and function of particular GABAergic neurons. Development 126, 1547–1562. Abstract
Huang, X., Powell-Coffman, J.A., and Jin, Y. (2004). The AHR-1 aryl hydrocarbon receptor and its co-factor the AHA-1 aryl hydrocarbon receptor nuclear translocator specify GABAergic neuron cell fate in C. elegans. Development 131, 819–828. Abstract Article
Ji, F., Kanbara, N., and Obata, K. (1999). GABA and histogenesis in fetal and neonatal mouse brain lacking both the isoforms of glutamic acid decarboxylase. Neurosci. Res. 33, 187–194. Abstract Article
Jiang, G., Zhuang, L., Miyauchi, S., Miyake, K., Fei, Y.J., and Ganapathy, V. (2005). A Na+/Cl- coupled GABA transporter, GAT-1, from Caenorhabditis elegans: structural and functional features, specific expression in GABAergic neurons, and involvement in muscle function. J. Biol. Chem. 280, 2065–2077. Abstract Article
Jin, Y., Hoskins, R., and Horvitz, H.R. (1994). Control of type-D GABAergic neuron differentiation by C. elegans UNC-30 homeodomain protein. Nature 372, 780–783. Article
Jin, Y., Jorgensen, E., Hartwieg, E., and Horvitz, H.R. (1999). The Caenorhabditis elegans gene unc-25 encodes glutamic acid decarboxylase and is required for synaptic transmission but not synaptic development. J. Neurosci. 19, 539–548.
Johnson, C.D., and Stretton, A.O. (1987). GABA-immunoreactivity in inhibitory motor neurons of the nematode Ascaris. J. Neurosci. 7, 223–235. Abstract
Kaupmann, K., Huggel, K., Heid, J., Flor, P.J., Bischoff, S., Mickel, S.J., McMaster, G., Angst, C., Bittiger, H., Froestl, W., and Bettler, B. (1997). Expression cloning of GABA(B) receptors uncovers similarity to metabotropic glutamate receptors. Nature 386, 239–246. Abstract Article
Keramidas, A., Moorhouse, A.J., French, C.R., Schofield, P.R., and Barry, P.H. (2000). M2 pore mutations convert the glycine receptor channel from being anion- to cation-selective. Biophys. J. 79, 247–259. Abstract
Keramidas, A., Moorhouse, A.J., Pierce, K.D., Schofield, P.R., and Barry, P.H. (2002). Cation-selective Mutations in the M2 Domain of the Inhibitory Glycine Receptor Channel Reveal Determinants of Ion-Charge Selectivity. J. Gen. Physiol. 119, 393–410. Abstract Article
Kravitz, E.A., Kuffler, S.W., and Potter, D.D. (1963). Gamma-aminobutyric acid and other blocking compounds in crustacea: III Their relative concentraitons in separated motor and inhibitory axons. J. Neurophysiol. 26.
Kuffler, S.W., and Eyzaguirre, C. (1955). Synaptic inhibition in an isolated nerve cell. J. Gen. Physiol. 39, 155–184. Article
Lee, R.Y., Sawin, E.R., Chalfie, M., Horvitz, H.R., and Avery, L. (1999). EAT-4, a homolog of a mammalian sodium-dependent inorganic phosphate cotransporter, is necessary for glutamatergic neurotransmission in Caenorhabditis elegans. J. Neurosci. 19, 159–167. Abstract
Liu, Y., Peter, D., Roghani, A., Schuldiner, S., Prive, G.G., Eisenberg, D., Brecha, N., and Edwards, R.H. (1992). A cDNA that suppresses MPP+ toxicity encodes a vesicular amine transporter. Cell 70, 539–551. Abstract Article
Mackenzie, B., and Erickson, J.D. (2004). Sodium-coupled neutral amino acid (System N/A) transporters of the SLC38 gene family. Pflugers Arch. 447, 784–795. Abstract Article
McIntire, S.L., Jorgensen, E., and Horvitz, H.R. (1993). Genes required for GABA function in Caenorhabditis elegans. Nature 364, 334–337. Abstract Article
McIntire, S.L., Jorgensen, E., Kaplan, J., and Horvitz, H.R. (1993). The GABAergic nervous system of Caenorhabditis elegans. Nature 364, 337–341. Abstract Article
McIntire, S.L., Reimer, R.J., Schuske, K., Edwards, R.H., and Jorgensen, E.M. (1997). Identification and characterization of the vesicular GABA transporter. Nature 389, 870–876. Article
Olsen, R.W. (1991). GABA and inhibitory synaptic transmission in the brain. Semin Neurosci. 3, 175–181. Article
Otsuka, M., Iversen, L.L., Hall, Z.W., and Kravitz, E.A. (1966). Release of Gamma-Aminobutyric Acid from Inhibitory Nerves of Lobster. Proc. Natl. Acad. Sci. USA 56, 1110–1115.
Roberts, E. (2000). Adventures with GABA: Fifty Years On. In: GABA in the nervous system: the view at fifty years, D.L. Martin, and R.W. Olsen, eds. (Philadelphia: Lippincott Williams & Wilkins), pp. 1–24.
Sagne, C., El Mestikawy, S., Isambert, M.F., Hamon, M., Henry, J.P., Giros, B., and Gasnier, B. (1997). Cloning of a functional vesicular GABA and glycine transporter by screening of genome databases. FEBS Lett 417, 177–183. Abstract Article
Schofield, P.R., Darlison, M.G., Fujita, N., Burt, D.R., Stephenson, F.A., Rodriguez, H., Rhee, L.M., Ramachandran, J., Reale, V., Glencorse, T.A., et al. (1987). Sequence and functional expression of the GABA A receptor shows a ligand-gated receptor super-family. Nature 328, 221–227. Abstract Article
Schuske, K., Beg, A.A., and Jorgensen, E.M. (2004). The GABA nervous system in C. elegans. Trends Neurosci. 27, 407–414. Article
Shan, G. Kim, K., Li, C., and Walthall, W.W. (2005). Convergent genetic programs regulate related motor neuron classes similarities and differences in Caenorhabditis elegans. Dev. Biol. 280, 494–503. Abstract Article
Takamori, S., Rhee, J.S., Rosenmund, C., and Jahn, R. (2000). Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature 407, 189–194. Abstract Article
Thomas, J.H. (1990). Genetic analysis of defecation in Caenorhabditis elegans. Genetics 124, 855–872.
White, J.G., Southgate, E., Thomson, J.N., and Brenner, S. (1986). The structure of the nervous system of Caenorhabditis elegans. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 314, 1–340.
Zhou, H.M., and Walthall, W.W. (1998). UNC-55, an orphan nuclear hormone receptor, orchestrates synaptic specificity among two classes of motor neurons in Caenorhabditis elegans. J. Neurosci. 18, 10438–10444. Abstract
*Edited by Joshua M. Kaplan. Last revised August 31, 2005. Published August 31, 2005. This chapter should be cited as: Jorgensen, E.M. GABA (August 31, 2005), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.14.1, http://www.wormbook.org.
Copyright: © 2005 Erik M. Jorgensen. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: [email protected]
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.