Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
Mutagenesis drives natural selection. In the lab, mutations allow gene function to be deciphered. C. elegans is highly amendable to functional genetics because of its short generation time, ease of use, and wealth of available gene-alteration techniques. Here we provide an overview of historical and contemporary methods for mutagenesis in C. elegans, and discuss principles and strategies for forward (genome-wide mutagenesis) and reverse (target-selected and gene-specific mutagenesis) genetic studies in this animal.
Mutations are the engine of natural selection and arise spontaneously through a myriad of mechanisms (Nei, 2007). In C. elegans, spontaneous point mutations, gene duplications, and deletions arise at rates of 2 × 10−8, 3.4 × 10−7, and 2.2 × 10−7 per gene per generation, respectively (Denver et al., 2004; Lipinski et al., 2011). These alterations to the genome result from replication and repair errors, endogenous oxidative damage, and exogenous environmental insults (Denver et al., 2009). Cosmic rays bombard the atmosphere of the Earth at a rate of approximately 2 × 1018 particles per second (Reedy et al., 1983), and they can also contribute to mutational load (Babcock and Collins, 1929). C. elegans has been used to study the effects of cosmic rays aboard the International Space Station (Zhao et al., 2006). Despite exposure to an average radiation dose of 10-12 mRad/day (approximately 150 times higher than exposure on the surface of the Earth (Ohnishi and Ohnishi, 2004)), no significant differences in mutation rates were detected over an 11-day period (Zhao et al., 2006), suggesting that DNA repair mechanisms must correct most mutations efficiently, and that spontaneous alterations to the genome may largely result from cell-intrinsic errors.
Mutations are important tools for gene function discovery. Forward and reverse genetic strategies rely on mutations to identify and characterize genes involved in a biological process of interest. A causal connection between mutation and phenotype can suggest mechanisms of wild-type gene action and can be used for diagnostic prediction. While gene function can be interrogated through targeted knockdown of RNAs using RNA interference (RNAi) (Fire et al., 1998), identification of genomic lesions in genes has several advantages. For example, the functions of genes that are not susceptible to RNAi can be assessed. Special alleles of genes whose inactivation leads to lethality, or that function redundantly, can also be identified (Muller, 1932). Temperature sensitive alleles allow for temporal assessment of gene activity (Hartwell et al., 1974), and single-gene mutations defining an allelic series can be used to study protein structure-function relationships (Jorgensen and Mango, 2002). Finally, while RNAi experiments sometimes give variable results (Kamath and Ahringer, 2003), genomic lesions tend to produce consistent phenotypic changes more suitable for quantitative analysis.
For spontaneous mutations to be an effective tool, roughly 1/(mutation rate) organisms must be screened to identify lesions in a particular gene. Often, one must screen well above this number. Although this is easily achieved in bacteria or yeast, the number is prohibitively high for other organisms, including C. elegans. Fortunately, the work of Muller (1927) demonstrated that mutation rates can be artificially enhanced by a variety of genotoxic agents. Practically, such treatments must maintain a balance between administration of sufficient damage (to reduce the numbers of queried animals) and animal lethality/sterility. In C. elegans, typical mutagenesis compatible with this balance inflicts 10−3 to 10−4 mutations per gene per generation (Gengyo-Ando and Mitani, 2000).
This chapter provides an overview of historical and current methods for gene mutagenesis in C. elegans. Three mutagenesis strategies are commonly used: genome-wide mutagenesis, in which unknown genes controlling a biological process are sought (forward genetics); target-selected mutagenesis, in which the entire genome is mutagenized but mutations in a known single gene are screened (reverse genetics); and gene-targeted mutagenesis, in which only a single gene is targeted for mutagenesis (reverse genetics). Method rationales and protocols are outlined, and are meant to serve as an introduction to aid researchers in identifying mutagenesis methods suitable to their goals (Table 1). Detailed protocols for each method are not presented for most methods, but are referenced and should be carefully followed when implemented in the laboratory.
Table 1. Overview of mutagenesis methods. Three types of mutagenesis are discussed in this review: whole-genome, target-selected, and gene-targeted mutagenesis. The different types and variations of each method are given; a general purpose and references for each technique are listed. Advantages and disadvantages of each method are discussed further in the text.
| Method | Types (references) | Purposes |
| Genome-wide mutagenesis | ||
| Chemical mutagenesis |
EMS (Brenner, 1974) TMP/UV (Barstead and Moerman, 2006) ENU (De Stasio and Dorman, 2001) formaldehyde (Johnsen and Baillie, 1988) NTG (Greenwald and Horvitz, 1980) DES (Greenwald and Horvitz, 1980) acetaldehyde (Greenwald and Horvitz, 1980) DEO (Anderson and Brenner, 1984) DEB (Trent et al., 1991) |
Forward genetic screens, target-selected mutagenesis |
| Radiation mutagenesis |
Short wave UV (Stewart et al., 1991) 32P decay (Babu and Brenner, 1981) |
Forward genetic screens, generate deficiencies |
| Transposon insertional mutagenesis |
Tc1 (Martin et al., 2002) Mos1 (Boulin and Bessereau 2007) |
Forward genetic screens, generate insertion mutants for use in gene-targeted mutagenesis |
| Target-selected mutagenesis | ||
| PCR-based methods |
restricted extension time (Jansen et al., 1997) poison primer (Edgley et al., 2002) thermostable restriction enzymes (Huang et al., 2006; Wei et al., 2002) |
Recover deletion in specific gene after whole genome mutagenesis |
| TILLING | (Gilchrist et al., 2006) | Isolate point mutations/allelic series in a gene of interest |
| G4 DNA-induced mutations | (Pontier et al., 2009) | Isolate deletion alleles for genes near G4 genomic site |
| Gene-targeted mutagenesis | ||
| Transposon-based methods |
Tc1 (Zwaal et al., 1993; Barrett et al., 2004) MosTIC (Robert, 2012) MosDEL (Frøkjaer-Jensen et al., 2010; Frøkjaer-Jensen et al., 2012) |
Make targeted changes or deletions in gene of interest without mutagenizing entire genome |
| Enzyme-based methods |
ZFNs (Wood et al., 2011) TALENs (Wood et al., 2011) CRISPR/Cas (Friedland et al., 2013; Dickinson et al., 2013) |
Create small indels or repair off a transgene to create mutations |
Non-directed genome-wide mutagenesis is commonly used for forward genetic screens, where mutants defective in a biological process of interest are sought. Mutagenesis can be accomplished with a variety of chemical agents, each offering a specific array of favored lesions and mutation frequencies. Ionizing radiation mutagenesis is used to generate large deletions or complex rearrangements. Alternatively, mutagenesis is achieved with transposon insertion, using either the endogenous transposons already present in the nematode or an exogenous Mos1 transposon.
Prior to embarking on genome-wide mutagenesis, a screen must be designed to identify mutant animals defective in the biological process under study. A thorough review of the various types of genetic screens in C. elegans has been previously published and should be consulted (Jorgensen and Mango, 2002). The most common screen involves identifying recessive mutations exposed in second-generation (F2) animals of a mutagenized parental strain (P0, Figure 1). Typically, P0 hermaphrodites are mutagenized at the late L4 or early adult stages. F1 progeny, heterozygous for many induced mutations, are then allowed to self, and F2 animals are screened for the phenotype of interest. Many variations on this screen exist, and each screen should be carefully constructed to find enough mutants for the phenotype of interest (Jorgensen and Mango, 2002). Because some genetic screens can be demanding in effort and time, it is prudent to optimize the ratio of F2-to-F1 animals assayed (Shaham, 2007). Calculations for the amount of effort required per screen center on whether F1 or F2 animals must be plated individually. We previously set up a web calculator to aid in evaluating optimal screen parameters (http://shahamlab.rockefeller.edu/cgi-bin/Genetic_screens/screenfrontpage.cgi). A detailed description of the theoretical considerations behind this calculator is available (Shaham, 2007). There are also techniques to automate and expedite a forward genetic screen, including the Complex Object Parametric Analyzer and Sorter (COPAS™), a device that sorts animals into individual wells, and microfluidic devices coupled to automated imaging (Chung et al., 2008; Crane et al., 2012; Doitsidou et al., 2008).
|
Figure 1. A simple F2 screen. Depicted here is the simplest variation of an F2 screen, which involves isolating mutants with a phenotype of interest in the second generation after mutagenesis. A population of young L4 hermaphrodites is mutagenized. F1 animals are collected, and F2 single hermaphrodites are isolated, carrying a homozygous mutation in a gene of interest. Obtaining a mutation is a rare event. The details of the scheme depend on the type of mutation sought. For a detailed description of different varieties of F2 screens, consult Jorgensen and Mango (2002).
Once a mutant is identified, several strategies can be used to isolate the affected gene. In the past, two- and three-point mapping with genetic markers was used to identify the affected region, although this method is not commonly employed today (for a review, see WormBook chapter Classical genetic methods). Single nucleotide polymorphism (SNP) mapping using the polymorphic Hawaiian isolate CB4856 is a more common approach (Wicks et al., 2001). Oligonucleotide array comparative genome hybridization (aCGH) has also been used successfully to identify point mutations and deletions in complex pools of genomic DNA (Maydan et al., 2007; Maydan et al., 2009). In this case, custom-made arrays with overlapping 50-mer probes are used to identify SNPs and insertions or deletions (indels). Whole-genome sequencing is now a cost-effective approach to gene mapping, and it can be combined with other mapping methods for rapid gene identification (Doitsidou et al., 2010; Sarin et al., 2008; Zuryn et al., 2010). There is open-source software to aid in whole-genome sequencing data analysis and mutant identification, including Bowtie, CloudMap, and galign, which are specifically tailored to the C. elegans genome (Langmead et al., 2009; Langmead and Salzberg, 2012; Minevich et al., 2012; Shaham, 2009).
Chemical mutagenesis provides the easiest and most straightforward way to induce germline mutations at high frequency. Mutation prevalence using chemicals is high enough that a relatively low number of haploid genomes must be interrogated to reach saturation. The most widely used mutagens in C. elegans are ethyl methanesulfonate (EMS) and trimethylpsoralen with ultraviolet light (UV/TMP). There are, however, many others with differing mutagenic spectra, including N-ethyl-N-nitrosourea (ENU), formaldehyde, nitrosoguanidine (NTG), diethyl sulfate (DES), acetaldehyde, diepoxybutane (DEB) and diepoxyoctane (DEO). A cocktail of EMS/ENU has also been used to induce a spectrum of mutations (Thompson et al., 2013). Special care should be taken when handling mutagens: working in a chemical hood and wearing proper personal protection gear are a must. Consult the relevant Material Safety Data Sheet (MSDS) and your chemical waste department before beginning a chemical genetic screen.
EMS is the most commonly used mutagen in C. elegans and by far the most potent (Brenner, 1974; Flibotte et al., 2010). A forward mutation rate as high as 2.5 × 10−3 mutations/gene/generation can be achieved with acceptable rates of sterility (Gengyo-Ando and Mitani, 2000). EMS is an alkylating agent and most commonly adds an ethyl group to guanine to form O6-ethylguanine (Brookes and Lawley, 1961). This modified guanine pairs inappropriately with thymine in subsequent replications (Coulondre and Miller, 1977). Therefore, mutagenesis is biased towards G/C to A/T transitions, respectively, which often leads to the generation of stop codons (Flibotte et al., 2010). Therefore, these mutations are typically strong loss-of-function or null alleles. Roughly 13% of EMS lesions are deletions or other chromosomal rearrangements (Anderson, 1995).
Protocol (Brenner, 1974):
Synchronized L4 or young-adult hermaphrodites are washed with M9 buffer in a sterile 15 ml plastic tube. Spin down animals at 700 × g for 1 min and remove supernatant. Wash animals at least 3x to remove bacteria. Resuspend animals in 2 ml M9. Add 2 ml of a 2x stock solution of EMS (100 mM). Animals are incubated in a total volume of 4 ml of 50 mM EMS in M9 for 4 h at 20oC with constant rotation. EMS should be prepared immediately before use. Animals are then washed 5x with 15 ml M9 to remove trace EMS. For standard F2 screens, plate mutagenized animals using a glass pipette on large NGM plates until F1 animals are gravid. Bleach F1 animals, and plate F2 eggs on NGM plates. Score for the defects of interest at the appropriate developmental stage. EMS is detoxified by addition of 1 M NaOH.
TMP is a light-sensitive interstrand cross-linking agent, whose mutagenicity is enhanced in the presence of long-wave UV light (Cole, 1970). UV/TMP generates small deletions with an average size of one to three kilobases; however, all base transitions and transversions have been observed experimentally (Flibotte et al., 2010; Gengyo-Ando and Mitani, 2000; Yandell et al., 1994). The forward mutation rate is approximately 1 × 10−3 mutations/gene/generation, but it depends on total UV exposure (Gengyo-Ando and Mitani, 2000).
Protocol (Barstead and Moerman, 2006):
Synchronized L4 animals are washed 5x in 15 ml M9 to remove bacteria. Animals are then resuspended in 580 µl M9 buffer. Twenty microliters of freshly made TMP (1 mg/ml in DMSO) is added to the culture, and animals are incubated in the dark for 15 min. Pipette animals onto dry, unseeded, 9 cm NGM plates. Remove plate lids, and apply 360 µJ UV light. A Stratalinker® UV Crosslinker (Stratagene) is useful for this step, but calibrated hand-held UV lamps can be used as well (Yandell et al., 1994). Next, add OP50 directly on top of the mutagenized worms and recover for 5 h in the dark at 20oC. Continue as for EMS screens. A range of final concentrations of TMP (2 µg/ml to 30 µg/ml) is reported to be successful. It is likely that variations depend on total UV exposure and variability between batches of TMP. Dose-response curves can be used to optimize mutagenesis for each batch (Barstead and Moerman, 2006).
ENU generates an array of molecular lesions, and it is therefore the mutagen of choice if allelic variability is important (Flibotte et al., 2010). The mutagenicity of ENU occurs by transferring its ethyl moiety to nitrogen or oxygen on any of the four nucleotides, creating modified DNA bases that later hinder proper replication (Goth and Rajewsky, 1972; Justice et al., 1999). All codons are equally mutable with ENU, so non-null alleles are often generated (De Stasio and Dorman, 2001). ENU has a slightly higher reversion frequency at around 2.6 × 10−4, compared to 1.1 × 10−4 using EMS (De Stasio and Dorman, 2001). The reversion frequency is the frequency at which a mutated phenotype reverts back to wild type. This is commonly a result of a second site suppressor mutation. Because there are usually fewer suppressor genes, this frequency is lower than the forward mutation rate (Greenwald and Horvitz, 1980).
Protocol (De Stasio and Dorman, 2001):
ENU degrades to more toxic products when stored in aqueous solutions (De Stasio and Dorman, 2001). Stock solutions of 50 mM in absolute ethanol should therefore be kept at -20oC for no more than 2 weeks. Here it is useful to buy small aliquots of dry ENU and add ethanol directly to the bottle to make the 50 mM solution (M. Edgley, personal communication). Working solutions (1.0 mM diluted in modified M9 buffer lacking ammonium chloride) are made fresh immediately before use (De Stasio and Dorman, 2001). Synchronized late L4/young adult hermaphrodites are incubated with the working solution for 4 h at 20°C with constant rotation. Animals are then washed 5x in M9 before plating on seeded NGM plates. Before discarding, ENU should be inactivated by adding sodium thiosulfate and sodium hydroxide to a final concentration of 10% (w/v) and 1% (w/v), respectively, and incubated at room temperature for 1 h.
EMS/ENU cocktail Protocol: The Million Mutation Project has used a cocktail of EMS and ENU to isolate mutations with a spectrum of lesions (Thompson et al., 2013). The same guidelines for each mutagen are used to prepare the mutagenic solution, and the final concentrations for mutagenesis are 50 mM EMS and 1 mM ENU in M9 (M. Edgley, personal communication). Synchronized L4/young adult hermaphrodites are washed into a 15 ml sterile tube. The animals are resuspended in 2 ml M9, and 2 ml of a 2x stock solution containing 100 mM EMS and 2 mM ENU are added. This solution is prepared fresh every time before use. The ENU stock solution (50 mM in absolute ethanol) is prepared and stored the same way as described for ENU mutagenesis.
The following mutagens are used less often, but they can also be considered for a forward genetic screen. The mutation rates given are rough estimates, as they are usually taken from only one or a few published screens. A thorough review of these uncommon chemical mutagens has been previously published (Anderson, 1995). In all cases, synchronized L4/young adult hermaphrodites are used. Following treatment, mutagenized animals are washed 5x in M9 buffer and plated on seeded NGM plates, according to the screen strategy.
Formaldehyde generates deficiencies, as well as point mutations and complex lesions, but it is less effective than EMS (Moerman and Baillie, 1981). Working concentrations of 0.1% (w/v) provide the best trade-off between mutagenic capacity and toxicity (Johnsen and Baillie, 1988). Stock solutions (1% (w/v)) are stored at −20°C in M9. Hermaphrodites are subjected to mutagenesis for 4 h at 20°C, rotating.
Nitrosoguanidine (NTG) is an alkylating agent that most likely causes point mutations by mispairing of bases during DNA replication (Lawley and Thatcher, 1970). It is used at a concentration of 0.5 mg/ml to mutagenize animals at a rate of 1 × 10−4 mutations per haploid genome (Greenwald and Horvitz, 1980). This rate is about ten times lower than that of EMS. In other organisms, however, NTG is a potent mutagen, suggesting that the best conditions for mutagenesis in C. elegans have not yet been determined (Anderson, 1995). Animals are incubated at 20°C for 1 h (Greenwald and Horvitz, 1980).
Diethyl sulfate (DES) is a mono-functional alkylating agent, and it has an approximate mutation rate of 5 × 10−4 mutations per haploid genome (Greenwald and Horvitz, 1980). Based on its chemical structure, it most likely causes point mutations (Brookes and Lawley, 1961). The precise spectrum of lesions, however, is unknown (Greenwald and Horvitz, 1980). Animals are incubated for 2 h in freshly made 1 mM DES diluted in M9 buffer at 20°C (Greenwald and Horvitz, 1980).
Acetaldehyde mutagenizes animals at a rate of approximately 1 × 10−4 mutations per haploid genome. Animals are incubated for 2 h at 20°C in 0.1% acetaldehyde in M9 (Greenwald and Horvitz, 1980).
1,2,7,8-diepoxyoctane (DEO) is a bi-functional alkylating agent that can generate large deletions, and it has an approximate mutation rate of 5 × 10−5 mutations per haploid genome. Hermaphrodites are incubated for 3 h in a freshly-made solution of 2 mM DEO in M9 (Anderson and Brenner, 1984).
1,2,3,4-diepoxybutane (DEB) has a similar reversion frequency to EMS. 15% of lesions are small deletions of 0.5-3.5 kb. Animals are incubated in 1 mM DEB in M9 buffer for 3 h at 20°C (Trent et al., 1991).
High-energy radiation mutagenesis is not commonly employed for contemporary forward genetic screens. Researchers should, however, consider such a method if large deletions or complex rearrangements are desired at a relatively high frequency (Muller, 1927).
Short-wave UV can induce chromosomal rearrangements, including deletions, translocations and duplications. DNA absorbs UV light, which causes adjacent pyrimidines to dimerize (Freeman et al., 1989). Lesions are then induced following inaccurate trans-lesion synthesis during replication (Bailly et al., 1994). UV mutagenesis has been used successfully to generate mutations at a rate of 2 × 10−4 mutations per haploid genome per generation (Greenwald and Horvitz, 1980). However, another study found that UV-C radiation had a low rate of mutation with a decrease in fertility, so more hermaphrodites should be used initially (Gengyo-Ando and Mitani, 2000). The reduction in brood size after irradiation was as high as 20% compared to unirradiated animals (Stewart et al., 1991). Young adult animals on uncovered, unseeded plates are irradiated with 120 W/m2 at 254 nm. Irradiated animals are allowed to expel eggs that were present prior to mutagenesis and are moved to new plates after 2 h (Stewart et al., 1991).
Ionizing radiation (IR) includes X- and γ-rays and generates very large deletions that may span several genes at rates of 2 × 10−4 to 9 × 10−4 (Anderson, 1995; Meneely and Herman, 1979; Sigurdson et al., 1984). IR also induces complex rearrangements including chromosomal duplications, translocations, and inversions. The lesions most likely differ from UV-induced lesions, as IR appears to have no nucleotide bias (Stewart et al., 1991). Synchronized L4/young adult hermaphrodites are irradiated with 1500 Roentgen and plated as needed. This method can recover non-lethal complex lesions (Rosenbluth et al., 1985). Alternatively, an F1 clonal screen can be used to isolate lethal alterations.
Sydney Brenner used 32P decay to generate some of the first C. elegans mutants (Babu and Brenner, 1981; Brenner, 1974). Mutations are generated at rates of 2.8 × 10−2 to 3.9 × 10−3 per haploid genome. 32P is incorporated uniformly into genomic DNA, and mutation is caused by 32P-transmutation in the genome, rather than by β-particle emission (Babu and Brenner, 1981). 32P is mixed with unlabeled phosphate and added directly to phosphate-free NGM plates at 10 Ci/mol (not a typo)! The plates are seeded with NA22, a radioactive-resistant strain of bacteria (Anderson, 1995). Freshly hatched larvae are added to the radioactive plates and grown at 15°C for 5 days. F1 animals are collected and plated on standard NGM plates seeded with OP50 as above (Babu and Brenner, 1981). As a considerable amount of radioactive waste is generated using this method, consult your institute's radioactive waste management facility prior to beginning.
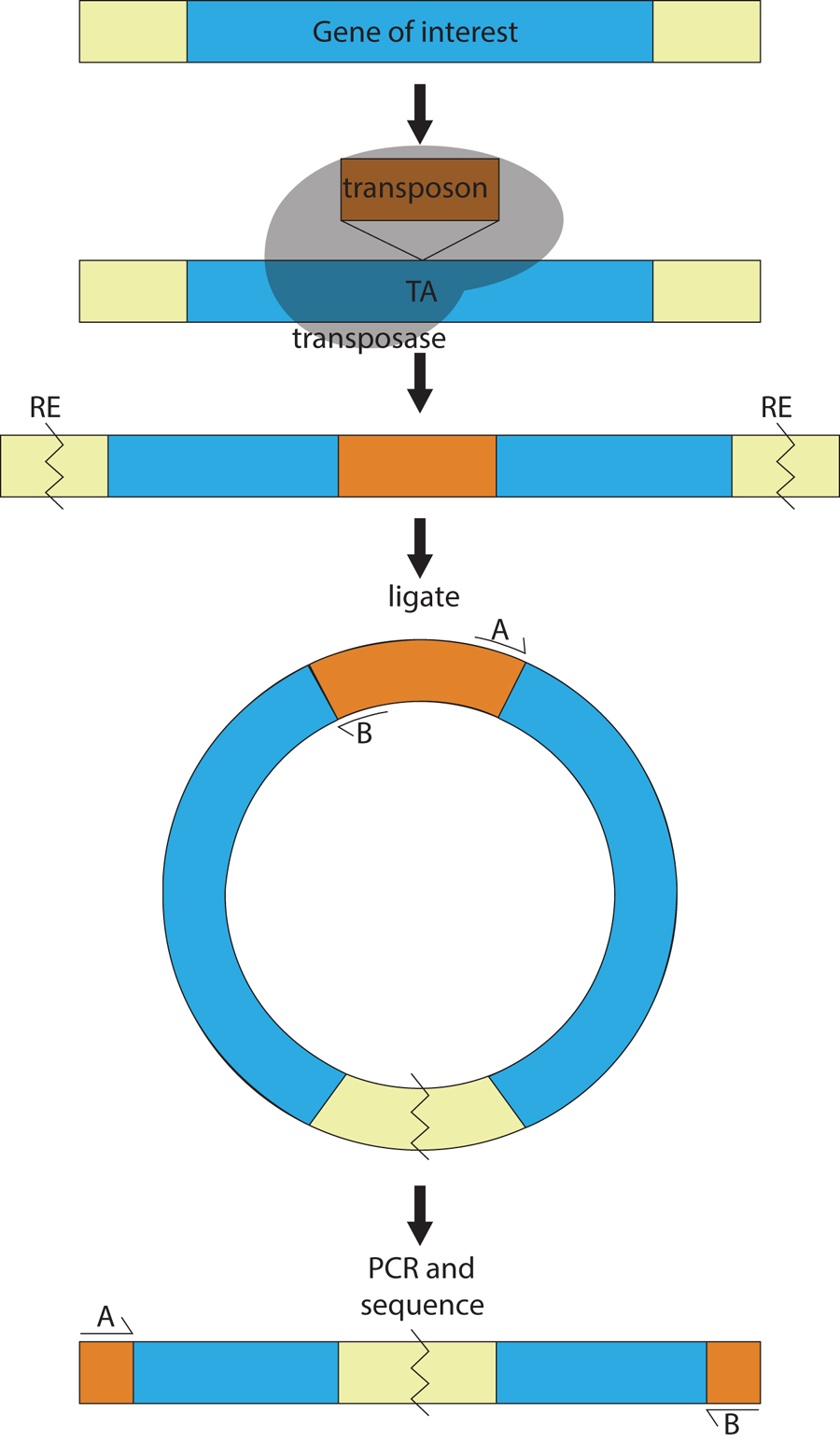
Transposons are DNA sequences that hop in and out of a genome with the help of a transposase enzyme, usually encoded by the transposon itself. In C. elegans, there are six active endogenous transposons, Tc1-Tc5 and Tc7 (Collins and Anderson, 1994; Collins et al., 1989; Emmons et al., 1983; Levitt and Emmons, 1989; Liao et al., 1983; Rezsohazy et al., 1997; Yuan et al., 1991). Tc1 is the most enriched transposon in the N2 strain, with approximately 30 copies (Emmons et al., 1983). Transposons are not normally active in the germline of N2, but they may become active when genes regulating RNAi and transposition are mutated (Collins et al., 1987; Vastenhouw et al., 2003). Fortunately, an exogenous transposon, Mos1, is active in the N2 germline in the presence of Mos1 transposase (Bessereau et al., 2001). Endogenous transposons and Mos1 insert randomly into the genome when activated in the appropriate background. Importantly, after insertion and following back-crossing, the transposon provides a molecular tag by which the disrupted gene can be identified using inverse (vectorette) PCR or whole-genome sequencing (Figure 2) (Smith, 2011; Williams et al., 2005). For a comprehensive review, consult Wormbook chapter, Transposons in C. elegans.
|
Figure 2. An insertional mutagenesis screen for isolating genes disrupted by a transposon. In insertional mutagenesis, transposons (Tc1, and more commonly Mos1) are mobilized in an appropriate strain background (mutator background or strain expressing Mos1-transposase). The transposon (orange) inserts randomly throughout the genome, via the transposase (gray), typically at a TA dinucleotide. DNA is isolated from animals that exhibit the phenotype of interest. Using inverse PCR, the DNA is cut with a restriction enzyme (RE) that is abundant throughout the genome and self-ligated. PCR is performed using two primers (A and B) within the transposon, elongating in opposite directions. PCR products (final panel) are size selected and sequenced to determine the position of insertion.
While generating mutations by chemical or radiation-induced mutagenesis is simple, identification of the relevant molecular lesion may not be. Major advances in SNP mapping and whole genome sequencing have made gene identification easier, yet these methods can be time consuming and labor intensive. This is especially the case when weakly penetrant or behavioral mutants are studied. In both cases, phenotype can only be established by scoring populations of animals, often requiring F3 or F4 animals to be evaluated. In these cases, non-directed insertional mutagenesis using transposons becomes a useful alternative.
There are several caveats to consider before performing a genome-wide screen using insertional mutagenesis. The Bristol reference strain N2 has 30 copies of Tc1 already present in the background, complicating identification of the causal lesion (Emmons et al., 1983). Both Tc1 and Mos1 have a bias for TA insertion sites; this makes transposons prone to inserting into introns, which may not inactivate the nearby gene or may be spliced out during transcription (Granger et al., 2004; Mori et al., 1988; Rushforth and Anderson, 1996). Mos1 has about a 10x lower mutation rate than EMS, meaning that ten times as many animals need to be scored to identify a phenotype of interest (Williams et al., 2005). Some mutations also occur as a result of ‘hit-and-run’ events, where transposon insertion and subsequent removal causes small indels to disrupt a gene (Boulin and Bessereau, 2007; Williams et al., 2005). These events greatly complicate gene identification. Because of these disadvantages, transposon-based insertional mutagenesis screening is used less often. Mos1-based methods are preferred. There are Tc1-derived alleles available from the CGC, so the descriptions below provide an overview of how those alleles were obtained.
C. elegans strains carry endogenous transposons, which have been used to perform a whole-genome insertional mutagenesis screen. Of the six endogenous, active transposons, Tc1 and Tc3 are screened most often, as these have the highest copy number in N2. All transposons, however, are active in most mutator backgrounds (Martin et al., 2002). Tc1 uses TA as an integration site, but with a relaxed target site specificity (Mori et al., 1988).
To activate transposition, special mutator strains are used. The most common mutator genes are mut-2, mut-7, and mut-6 (Collins et al., 1987; Ketting et al., 1999; Tabara et al., 1999). rde-2, mut-14, and mut-16 are also available (Mori et al., 1988; Tijsterman et al., 2002; Vastenhouw et al., 2003). Because these mutator strains have high rates of transposon hopping, they tend to accumulate mutations quickly, and they should be freshly thawed from frozen stocks rather than propagated on plates (Anderson, 1995).
Protocol (Martin et al., 2002):
Prior to screening, a mutator strain is backcrossed four times to the strain that will be mutagenized. Outcrossing limits the number of Tc1 copies already present in the mutator strain and removes any extraneous mutations prior to screening. Animals are allowed to grow for ten generations to accumulate mutations and are then screened for defects of interest (Martin et al., 2002). Once the phenotype is confirmed, strains are immediately outcrossed to the parental strain. This removes the mutator locus and prevents transposon excision from the gene of interest. Transposon insertion sites are identified similar to Mos1 identification below. Since there are multiple copies of Tc1 in the background, SNP mapping is helpful in determining the chromosome of interest and reducing the possible number of candidate genes. As is the case for Mos1, most insertions occur in intergenic regions and introns, but a small percentage also appear in exons (van der Linden and Plasterk, 2004).
Mos1 was originally isolated from Drosophila mauritiana (Jacobson et al., 1986). The consensus insertion site is the dinucleotide TA, in the context of an ATATAT sequence. Mos1, however, can insert in sequences lacking the consensus site. 50% of insertions map near or within genes (Granger et al., 2004). Intron insertion bias is observed, presumably because these tend to be T/A rich; however, 20% of insertions still occur within coding regions (Granger et al., 2004). Mutagenesis rates are an order of magnitude lower than chemical mutagenesis. As a result, ten times as many F1 animals must be screened to identify lesions in a given gene (Williams et al., 2005). It is advisable to start with a small pilot screen using EMS first. This determines an approximate number of mutants expected and ensures that an insertional screen is feasible (Boulin and Bessereau, 2007). Additionally, 6-15% of transposon-induced mutations arise through a ‘hit-and-run’ mechanism, in which the transposon imprecisely excises before the transposase becomes inactive (Boulin and Bessereau, 2007; Williams et al., 2005). On average, 2.5 insertions are generated per line, but as many as ten insertions in a single strain have been detected (Boulin and Bessereau, 2007). Mos1 has a length of 1.3 kb, so alleles generated tend to be strong loss-of-function. Hypomorphs can be generated if the transposon is inserted into regulatory sequences (Boulin and Bessereau, 2007).
Protocol (Boulin and Bessereau, 2007):
The Mos1 transposase and transposon array are maintained as extrachromosomal arrays in two different strains (EG2762 and EG1470). These are maintained as separate stocks to block spontaneous transposon insertions. EG2762 carries the Mos1 transposase under the control of a heat-inducible promoter, and a coelomocyte GFP marker. EG1470 has a pharynx GFP marker and the Mos1 transposon. Immediately prior to the start of the screen, the two strains are crossed to generate doubly transgenic animals. After one generation, the doubly transgenic strain is split into two populations. One population is used to test transposition frequency following heat shock to ensure that silencing of the transposon array in the germline has not occurred. The other is amplified for an additional 2-4 generations. Once transposition has been confirmed, the amplified doubly transgenic animals are subjected to a heat pulse in a 33°C water bath for 1 h, followed by 1 h incubation at 20°C, followed by another 1 h heat shock. Animals are recovered at 20°C for 12 h, transferred onto fresh plates and grown at 20°C. F1 progeny are picked 24 h to 36 h after heat shocks. For a comprehensive protocol and useful tips, see (Boulin and Bessereau, 2007).
Target-selected mutagenesis is used to identify mutations in a specific gene from a randomly mutagenized genome. The approach involves standard mutagenesis using EMS or UV/TMP, for example, followed by screening using the polymerase chain reaction (PCR) or other method to identify animals carrying lesions in the gene of interest.
Two community consortia, the C. elegans Knockout Consortium (celeganskoconsortium.omrf.org), and the C. elegans National Bioresource Project of Japan (NBRP, http://www.shigen.nig.ac.jp/c.elegans/mutants/index.jsp) are currently using target-selected mutagenesis to identify mutant alleles for every gene in the C. elegans genome (C. elegans Deletion Mutant Consortium, 2012). Together, over 6,700 deletion or null alleles have been isolated by these groups and are available to the scientific community (C. elegans Deletion Mutant Consortium, 2012). Strains from the C. elegans Knockout Consortium are available from the Caenorhabditis Genetics Center (CGC, http://www.cbs.umn.edu/research/resources/cgc) under the allele designation ok or gk. Strains generated by the NBRP, with allele designation tm, are available after submission of a Materials Transfer Agreement (Mitani, 2009). Deletion strains are listed on Wormbase, which should be consulted first to determine if an allele is already available for a gene of interest. If not, one may request a deletion strain from the Consortium. Upon receiving a strain, it should be outcrossed to remove irrelevant mutations. It is always important to verify the presence of the mutation in a strain. In addition, deletion mutations obtained by target-selection can be accompanied by proximal duplications, resulting in obligate heterozygosity for the gene under consideration. In these situations, phenotypic assessment of the mutant strain may not reveal the defects associated with complete gene loss. To confirm that a gene of interest is responsible for an observed defect, transgenic rescue experiments should be performed and/or multiple alleles examined (C. elegans Deletion Mutant Consortium, 2012). In addition to the collection of deletions, the Million Mutations Project (http://genome.sfu.ca/mmp/) has identified over 800,000 single nucleotide polymorphisms (SNPs) and over 16,000 indels in more than 20,000 genes (C. elegans Deletion Mutant Consortium, 2012; Thompson et al., 2013). While some of these SNPs do not affect gene function, many may provide useful structure-function data.
The goal of target-selected mutagenesis is to generate a library containing pools of mutagenized animals that can be screened for specific DNA lesions. Deletion alleles are easier to identify using PCR than point mutations, so UV/TMP and EMS are the preferred mutagens (Lesa, 2006). For library construction, 600,000 hermaphrodites are randomly mutagenized. Once the mutagenized animals become gravid, they are bleached and L1s are allowed to hatch in M9, overnight. The L1s are then subdivided into 1,152 subcultures (55 mm NGM plates) of 500 animals each. After two generations of self-fertilization, 20% of the animals per plate are rinsed off for genomic DNA isolation into twelve 96-well plates (Lesa, 2006). The remaining animals are stored at 15°C for up to six weeks. Although libraries can be frozen, live-animal libraries are less prone to failures in mutant recovery (Lesa, 2006).
Three approaches are commonly used to search for deletions in a gene of interest: restricted PCR conditions, poison primers, and thermostable restriction enzymes. Each approach employs nested PCR primers designed to flank the genomic sequence of interest to increase specificity, and each method favors amplification of deletion products over larger wild-type products (Edgley et al., 2002; Jansen et al., 1997; Wei et al., 2002; Zwaal et al., 1993). All of these methods are prone to high false positive rates (50%-90%); therefore, each pooled sample should be screened in duplicate (Jansen et al., 1997). A comprehensive protocol for library creation and the poison primer PCR technique is available in the Reverse genetics chapter in WormBook.
Each library pool is incubated first with a primer pair (10 pmol per primer) designed 2.5-3.5 kb apart over a region of interest, followed by incubation of a 1:500 dilution of the first PCR with a nested primer pair (Liu et al., 1999). After addition of polymerase, nucleotides and buffer, samples are cycled with short extension times (45 s to 1 min) to favor accumulation of the deletion product (Jansen et al., 1997). DNA polymerases have specific turnover rates (maximum number of nucleotides polymerized in a given amount of time), which vary depending on the type of Taq used (Kornberg and Baker, 1992). For example, if the wild-type (WT) product is 2 kb, the deletion product is 500 bp, and the Taq polymerase has a turnover rate of 1 kb/min, approximately two min will be needed to complete the WT reaction and 30 sec for the deletion product. If the extension time is limited to 45 s, only the deletion product will be amplified. Over many rounds, some WT product is amplified, but at a much slower rate than the deletion. This greatly increases the odds of detecting the less abundant deletion allele. Detected deletions are variable in extent and average around 1.4 kb in size (Liu et al., 1999). Completed reactions are run on an agarose gel, and pools that have a product smaller than the wild-type product are identified. Library animals from this pool are plated either singly or in smaller pools and retested to identify single hermaphrodites carrying the relevant lesion (Jansen et al., 1997).
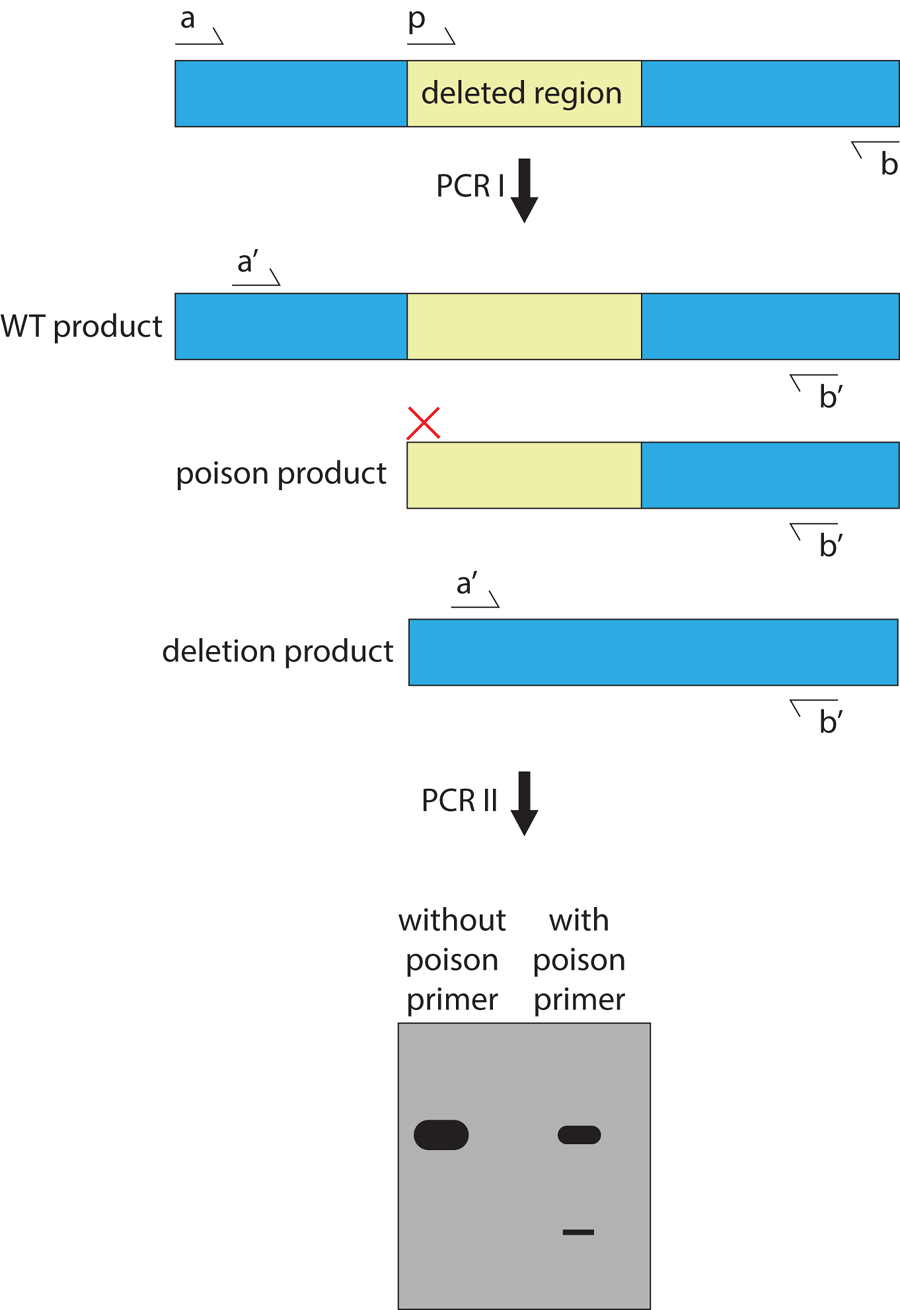
This approach was developed to enrich for deletions that are less than 500 bp in size (Edgley et al., 2002). The protocol is similar to that for the restricted PCR extension time method, except that a third primer is added between the external primers of the first PCR (Figure 3). This poison primer is designed to amplify only off of wild-type genomic DNA, competing with the full-length wild-type product, thereby reducing the quantity of wild-type template available for the second PCR. The second set of nested primers is only able to bind the deleted product and the full-length wild-type product, whose total concentration is halved because of the poison primer product. This leads to a 500-fold increase in sensitivity by enhancing the ratio of deletion to wild-type products. Use of a short extension time (45 s) also facilitates synthesis of the deletion product. Because the poison primer determines the location of amplified deletion products, it is usually best to target the 5’ end of a gene to generate null alleles (Edgley et al., 2002).
|
Figure 3. Poison primer method for detecting deletions. This technique enriches rare, deleted DNA over the more abundant, full-length, wild-type product. Primers a, b, and p are used in the first PCR to generate a wild-type template, a mutant template, and a ‘poison’ template. In the second PCR, these products are used as template with primers a’ and b’ to increase the signal of the deleted product. Without the poison primer, full-length product dominates the PCR reaction, and the deletion product is below the detection limit. When the poison primer is included, total full-length amplification is decreased, and the deletion product is revealed.
In this less commonly used method, sequences in a target gene are identified that have a thermostable restriction enzyme recognition site in the interval spanned by the primer pairs (Wei et al., 2002). These enzymes are stable at high temperatures, making them ideal to use with PCR. Pooled genomic DNA is digested for 2 h before PCR to cleave genomic DNA and reduce the amount of wild-type template available. The enzymes are also included during the PCR amplifications to continuously cleave any wild-type product. Two sets of nested primers are used. Targeted deletion sequences lack the restriction enzyme site and are not cleaved, thereby increasing the sensitivity of the reaction (Wei et al., 2002). Deletions ranging from 330 bp to 1.7 kb have been found using this method (Huang et al., 2006; Wei et al., 2002). Validated thermostable restriction enzymes include PspGI, TliI, BstUI, ApeKI, and Tsp45I (Huang et al., 2006; Wei et al., 2002).
TILLING (Targeting Induced Local Lesions IN Genomes) allows the identification of point mutations and small indels in a gene of interest (McCallum et al., 2000). Originally developed in Arabidopsis thaliana, TILLING uses a single-strand DNA nuclease to determine the location of mismatches after hybridizing a mutated genome to a non-mutated genome (Colbert et al., 2001; Till et al., 2003). While this method has only been used once to isolate point mutations in a gene of interest in C. elegans, it has been used more in other organisms. Mutations in a population of C. elegans are generated using EMS or ENU (Gilchrist et al., 2006). PCR targeting a gene of interest is performed on genomic DNA derived from many pools of animals (Figure 4). Primers are tagged with different fluorophores, marking the 5’ and 3’ ends of the product with different colors. After PCR, the DNA in each reaction is denatured. Reannealing then allows heteroduplex formation between wild-type and mutant DNA. Reannealed DNA is incubated with a single-strand DNA nuclease, CEL1, derived from celery juice extract (CJE) (Till et al., 2004). CJE cuts heteroduplex DNA where a mismatch or indel creates a single-stranded bulge. Samples are run on denaturing LI-COR gels, and the gels are examined in both fluorescent channels to determine in which pool a mismatch occurs (Gilchrist et al., 2006). In a screen for ten genes (ranging from 788 bp to 9.1 kb in size), 71 mutations were identified. Of these, 59% constituted missense alleles, and 3% were nonsense alleles (Gilchrist et al., 2006).
|
Figure 4. TILLING allows identification of point mutations in pools of mutated DNA. Both mutated DNA and wild-type DNA are subjected to PCR, using differentially labeled 5’ and 3’ primers (here in green and red). The PCR products are denatured and reannealed slowly, so that heteroduplex DNA forms between a mutated DNA strand and a wild-type DNA strand. The reannealed PCR products are treated with Cel1, an enzyme that specifically cleaves heteroduplex DNA. The reaction is visualized on a LI-COR gel in two fluorescence channels. When heteroduplex DNA is cut, smaller DNA fragments are seen in both the green and red channels within the same lane. Full-length, uncut product will be prominent at the top of the gel.
Protocol (Gilchrist et al., 2006):
Individual mutagenized F1 animals are plated singly onto 1500 seeded NGM plates. After the food supply is expended, half of the animals are frozen at −80°C. The other half are used to generate genomic DNA individually into 96-well plates. To reduce the number of PCR reactions, eight F1 DNA samples are pooled together for analysis. Once a positive pool has been identified, DNA genomic samples are assayed individually to determine from which plate the sample originally arose. PCR primers are designed over an area of 1.5 kb, mainly encompassing exonic regions. Primers can be designed using the program CODDLE to select for regions high in G/C content, which are most prone to mutation by EMS (Gilchrist et al., 2006).
The consensus sequence G3+N1-7G3+N1-7G3+ N1-7G3+ folds in vitro to form a guanine quadruplex (G4) structure that may block replication in vivo (Kruisselbrink et al., 2008). The FANCJ protein (DOG-1 in C. elegans) is a DNA helicase required for proper replication of such guanine-rich tracts (Kruisselbrink et al., 2008). In dog-1 mutants, these tracts are prone to 5’ deletions. There are more than 2,900 guanine quadruplex sites in the C. elegans genome, annotated in Wormbase under “G4 Motif” in GBrowse, and over 1,600 genes are located 5’ of a G4 site (Pontier et al., 2009). Of ten genes assayed in one study, 11 deletion alleles were generated, most of which were up to several kilobases upstream of the G4 site targeted (Pontier et al., 2009). This method has been used once to generate deletion alleles (Pontier et al., 2009).
Protocol (Pontier et al., 2009):
dog-1(pk2247) mutants are picked to individual NGM plates and allowed to grow until no OP50 remains on the plate (Pontier et al., 2009). Half of the animals on each plate are rinsed off with M9 for genomic DNA isolation. Prior to DNA isolation, animals are split into two groups. If a deletion is present in only one of the samples, it probably arose from a somatic event. However, if a deletion is detected in both samples, it is most likely a heritable mutation (Pontier et al., 2009). Deletions are screened using any of the PCR protocols described above. After isolation, the strain is outcrossed to wild-type animals to remove the dog-1 mutation and any off-target deletions.
Gene-targeted mutagenesis methods seek to generate lesions in a specific gene of interest without mutagenesis of the entire genome. Nonetheless, strains generated with these methods should still be outcrossed before performing genetic studies, as off-target lesions can occasionally occur. In general, however, the mutational load is much lower than that inflicted with chemical or radiation mutagenesis.
Transposon-mediated, transgene-directed gene conversion using Tc1 can generate specific alleles in a target gene, albeit at lower frequencies than Mos1 (Bessereau et al., 2001). Tc1-based methods are not commonly used anymore with the advent of single insertion Mos1 strains. Some historic alleles were obtained using Tc1, so the following protocol serves as reference for how those alleles were generated. Tc1-based methods require that a transposon is located near or in the gene of interest. Once such a strain is identified, one can either screen for excision events that remove part of the gene, or introduce a mutated transgene to direct gene conversion.
Protocol:
For isolating deletion events, animals containing a Tc1 insertion in the gene of interest are crossed into a mutator background (e.g., mut-2), and at least 100 animals are individually plated and allowed to self for two to three generations (Zwaal et al., 1993). Half of the animals on each plate are washed off for DNA isolation, and screened for deletions using PCR. If a deletion is detected, individual animals from the remaining half are plated individually and assayed for the presence of the deletion. Typical excision rates are 10−3 per gene per generation (Kagan et al., 1997). Once a deletion has been identified, the strain is outcrossed several times to the wild-type strain to remove mut-2 and any extraneous mutations.
For transgene-directed gene conversion, the protocol is similar, except that a transgene with homology to sequences flanking the transposon insertion site is injected into the gonad first. The transgene is engineered with a point mutation, deletion or GFP insertion (Barrett et al., 2004; Plasterk and Groenen, 1992). Progeny are then screened for homologous recombination from the engineered transgene by PCR. Gene deletions of 0.85 kb to 1.5 kb have been targeted successfully in a mut-2 background, with a frequency of 1/2666 animals undergoing gene conversion with a small deletion template (Barrett et al., 2004).
Mos1 excision-induced Transgene-Instructed gene Conversion (MosTIC) introduces specific point mutations, small deletions, and insertions (e.g., of GFP) into a C. elegans gene of interest (Robert and Bessereau, 2007). A strain containing a genomic Mos1-insertion near or in a target gene is identified. The NemaGENETAG Consortium created a collection of over 13,000 Mos1-tagged alleles (Bazopoulou and Tavernarakis, 2009; Vallin et al., 2012). The Mos1-insertion strains are annotated on MosLocator (http://www.ciml.univ-mrs.fr/applications/MosLocator/) and Wormbase, and they are available to researchers through the Consortium (http://ums3421.univ-lyon1.fr/). Once the strain of interest is received, it is transformed with a transposase used to remobilize the transposon and with a transgene containing a repair template with a specific mutation in the region of the Mos1 insertion. Excision of the Mos1 element following transposase expression can then be repaired by homologous recombination with the provided repair template, thus introducing specific lesions into the genome. Gene conversion events occur at approximately 2 × 10−4 events per F1 animal isolated, so one would need to screen at least 5000 animals to isolate one successful conversion event (Robert and Bessereau, 2007). A similar strategy, MosDEL, can be used to introduce large deletions at specific sites (Figure 5) (Frøkjaer-Jensen et al., 2010). Deletions of up to 25 kb have been generated using the MosDEL method, at efficiencies of approximately 20% of injected worms, such that in 30 positively-selected, transgenic worms, six will carry the targeted deletion (Frøkjaer-Jensen et al., 2012). 99.4% of all genes in the C. elegans genome are within 25 kb of at least one Mos1 element in strains generated by the NemaGENETAG, with a median distance of 3.1 kb, essentially making all genes targetable by this method (Frøkjaer-Jensen et al., 2010). The related MosSCI method, which also uses homologous repair after excision, can be used to introduce single copies of a transgene into a specific genomic site, but it is not a mutagenic technique (Frøkjaer-Jensen et al., 2012; Frøkjaer-Jensen et al., 2008).
|
Figure 5. Targeted gene deletions using transposons. To generate a targeted gene deletion, a transposase and a rescuing transgene containing a selection marker (in this case, unc-119) is injected into an unc-119 mutant animal carrying a transposon insertion near or in a gene of interest. Typically, strains carrying a single Mos1 insertion near a gene of interest are obtained from the NemaGENETAG Consortium (Bazopoulou and Tavernarakis, 2009; Vallin et al., 2012). Homology-directed repair after a double-strand break (DSB) occurs off the introduced transgene at low frequencies. unc-119(+) is inserted into the genome, and in the case of MosDEL, some portion of the gene is deleted, typically the 5’ region of the gene. In MosTIC, the transgene contains a small insertion, deletion, or point mutation, which is incorporated into the genomic DNA.
Protocol:
MosTIC: (Robert, 2012) The transgene for repair is designed to contain point mutations, small deletions, or insertions in a region spanning about 1 kb around the Mos1 insertion site. These modifications should be flanked by at least 1.5 kb of genomic sequence homology to facilitate recombination. This repair transgene (50 ng/µl) is introduced into the Mos1-containing strain via DNA microinjection, along with a hsp::Transposase plasmid (50 ng/µl) and a co-injection marker (Robert, 2012). After a line is established, approximately 200 transgenic animals undergo Mos1 excision for 1 h at 33°C. Animals are allowed to recover for 1 h at 15°C, and then shifted once more for 1 h to 33°C. After recovery overnight, bleach gravid hermaphrodites and collect the F1s. Plate 100 F1s per plate over many NGM plates and allow to self-fertilize for 2 generations (Robert, 2012). Alternatively, one to five heat-shocked P0 animals may be plated to generate 100 F1 animals per plate (Robert and Bessereau, 2007). After, proceed with a PCR screening strategy as above (Section 4.1.1). One may also screen by phenotype using a different plating strategy if the phenotype is known. The gene conversion event should still be confirmed by PCR, as false positives may arise (Robert, 2012). Expression of the transposase directly under a germline promoter (eft-3) rather the heat shock promoter increases efficiency, using a protocol similar to that for MosDEL outlined below (Frøkjaer-Jensen et al., 2012; Robert, 2012).
MosDEL: (Frøkjaer-Jensen et al., 2010) The overall protocol for MosDEL is similar to that for MosTIC. Targeting transgenes used for MosDEL should have one homology arm containing 2 kb of DNA directly adjacent to one side of the Mos1 insertion site. The other homology arm should have at least 3 kb of DNA targeting sequence upstream, chosen to mark the end point of the deletion (Frøkjaer-Jensen et al., 2010). Within the homology arms, GFP, C. briggsae unc-119(+), or an antibiotic-resistance gene can be inserted as a positive selection marker (Frøkjaer-Jensen et al., 2012; Giordano-Santini et al., 2010; Semple et al., 2010; Semple et al., 2012). One advantage of unc-119(+) is that it can act as a balancer in case the deletion results in lethality when homozygous. Also, unc-119 mutant animals are unable to form dauers and die in starvation conditions, which further aids screening (Maduro and Pilgrim, 1995; Praitis et al., 2001). The C. briggsae unc-119 sequence is used because it is smaller than the C. elegans coding region, but it still rescues unc-119 mutants (Frøkjaer-Jensen et al., 2010). The injection mix should contain the repair plasmid (50 ng/µl), a Mos1 transposase under the eft-3 promoter to drive germ line expression (50 ng/µl, (Frøkjaer-Jensen et al., 2012)) and co-injection markers (at least three different, well-expressed markers to reduce false positives) (Frøkjaer-Jensen et al., 2012). Deletion mutants are isolated by picking unc-119(+), GFP(+), or antibiotic-resistant animals that do not express the co-injection markers. A negative selection marker such as peel-1 may be included as a co-injection marker to increase the efficiency of screening after a Mos1 event (Frøkjaer-Jensen et al., 2012). peel-1 is placed under a heat-shock promoter, and is induced for 2 h at 34°C. Transgenic animals carrying the array are killed, while animals that have lost the transgene survive. This selection will aid in screening for animals that lost the co-injection markers but repaired off the transgene successfully (Frøkjaer-Jensen et al., 2012). Deletions are confirmed by PCR and sequencing. The repair template may be engineered to contain Flp recombinase sites, so that the positive selection marker may be removed in somatic cells to create a clean deletion (Vazquez-Manrique et al., 2010).
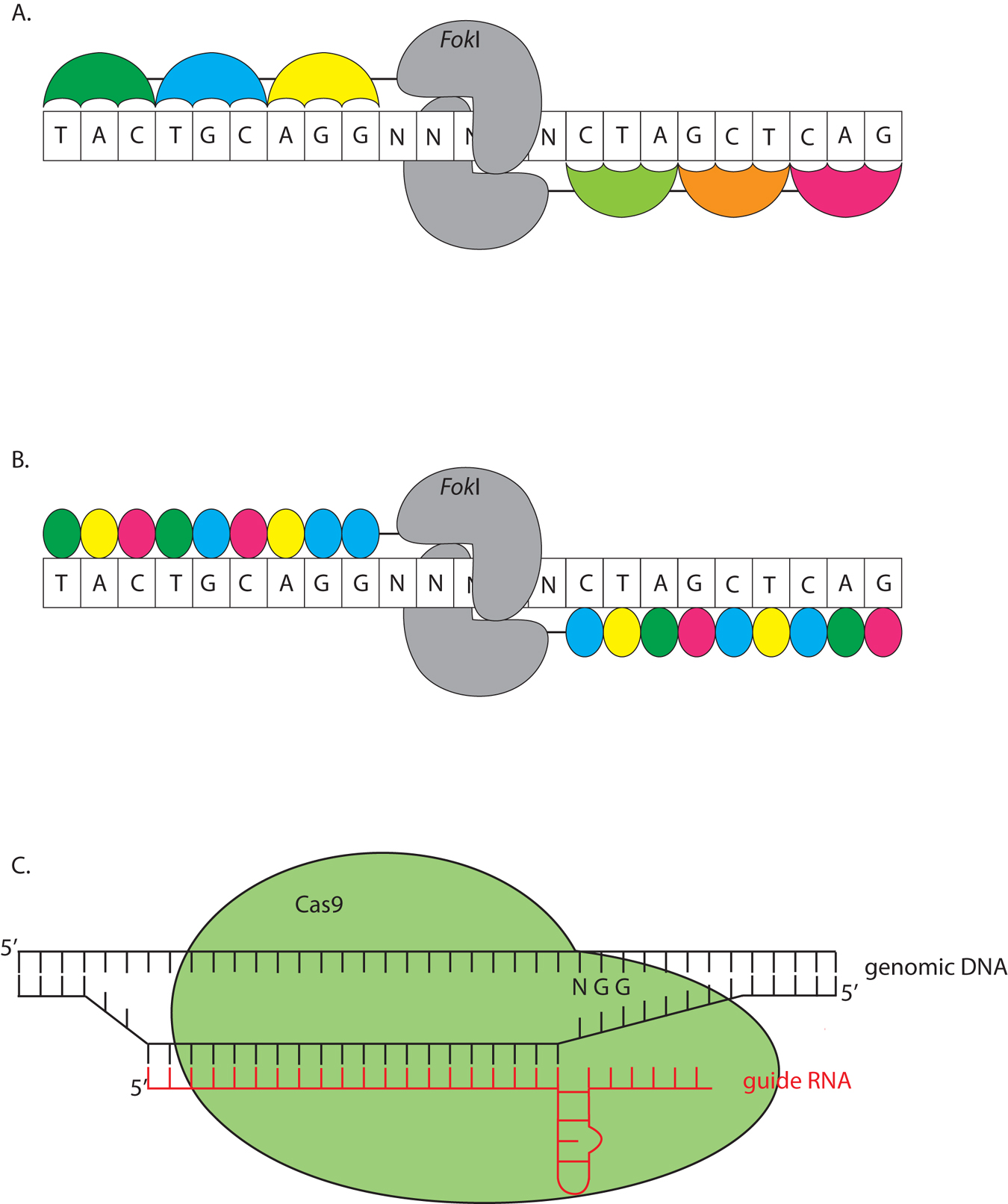
Zinc finger nucleases (ZFNs) are engineered chimeric proteins, consisting of tandem zinc finger domains fused to a modified FokI restriction enzyme (Figure 6A) (Kim et al., 1996). The zinc finger domains are engineered to recognize specific sequences. FokI cleaves DNA in the vicinity of the binding site. Typically, three zinc finger domains, recognizing a 9-nt sequence, are fused to FokI. Two such proteins are generated (with a combined recognition sequence of 18 bp) allowing dimeric FokI to cleave DNA (Kim et al., 1996). Faulty repair occasionally introduces small indels to disrupt a targeted gene. ZFNs require considerable functional tailoring; therefore, they are expensive to generate.
|
Figure 6. Molecular scissors can be used to engineer small insertion and deletions into a genome. A. Zinc finger nucleases (ZFNs) consist of three tandem Zinc finger domains (colored umbrellas) fused to a FokI nuclease (gray). FokI acts as a dimer, and specifically cleaves genomic DNA when two ZFNs are targeted in tandem. B. TALENs are similar to ZFNs, except that a single domain recognizes a single nucleotide, so one of four domains is fused together according to the genomic sequence. The nuclease is traditionally FokI (gray), functioning as a dimer, although monomeric TALENs using I-TevI have recently been reported (Beurdeley et al., 2013). C. The CRISPR system uses a single guide RNA (red) to target a region of interest for cleavage. The Cas9 nuclease (green) recognizes aspects of the secondary structure of the single guide RNA and the RNA:DNA hybrid. Cas9 also requires a protospacer motif, NGG, to cleave the DNA.
TALENs function like ZFNs, except that tandem TAL domains are fused to a FokI protein (Figure 6B). TAL domains allow a much greater freedom in choosing a DNA sequence to target (Miller et al., 2011). Each domain is tailored to a specific nucleotide, rather than a tri-nucleotide sequence. The TAL code is modular (Boch et al., 2009; Moscou and Bogdanove, 2009), allowing targeted nucleases to be generated at lower cost using Golden Gate cloning, which compiles multiple DNA fragments in an ordered fashion by traditional cloning techniques (Cermak et al., 2011). An improvement to the system has been reported by fusing a single TAL domain to the catalytic domain of an I-TevI homing endonuclease, allowing the enzyme to function as a monomer (Beurdeley et al., 2013). Although not yet used in C. elegans, it may allow TALEN mRNA to be injected at lower concentrations and be more effective (see Protocol below).
In prokaryotes, the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated (Cas) system is an adaptive immune response against viruses and plasmids (reviewed in Wiedenheft et al., 2012). The cell uses an RNA guide to cleave foreign DNA sequences (Figure 6C). The minimum components required to obtain cleavage in vitro for the Type II CRISPR system are the Cas9 nuclease from Streptococcus pyogenes and a synthetic single guide RNA (sgRNA) composed of a short CRISPR RNA (crRNA) and a trans-activating CRISPR RNA (tracrRNA) (Jinek et al., 2012). Genomic specificity is provided by a 20-bp sequence within the crRNA, whose only constraint is the trinucleotide NGG (Protospacer-Adjacent Motif or PAM) after the 3′ end of the sequence. In genomic editing, after successful targeting by the sgRNA, Cas9 introduces a double-strand break 3 bp upstream of the PAM site, which may introduce indels after error-prone non-homologous end joining (NHEJ) or can repair off an introduced transgene using homology directed repair (HDR). To promote HDR over NHEJ, Cas9 can be converted into a nickase (Cas9n) by a single amino acid substitution (Cong et al., 2013; Mali et al., 2013). Off-target lesions have been recovered in studies using human cell culture, and single and double mismatches appear to be tolerated in some systems (Fu et al., 2013; Cradick et al., 2013). While no off-target effects have been reported for studies in C. elegans (Dickinson et al., 2013; Chiu et al., 2013), it is good practice to backcross the mutant animal to remove any possible undesired lesions. To increase fidelity and reduce off-target hits, two Cas9n enzymes can be used together to target a genomic locus (Ran et al., 2013). One Cas9n nicks the sense strand, while the other nickase targets the anti-sense strand, with an offset of the two sgRNAs between 0 and 20 bp. Double nicking creates 5′ overhangs that may be repaired by NHEJ and introduce lesions. This method was found to greatly decrease off-target lesions in mice zygotes and human cell culture by 50 to 1500 fold (Ran et al., 2013). Additionally, because of the small size of the targeting constructs, multiple loci can be edited at once (Cong et al., 2013).
When ZFNs and TALENs were initially tried in C. elegans, germline cleavage was difficult to obtain: ZFN-directed cleavage only occurred in somatic cells (Morton et al., 2006). However, a recently developed protocol uses mRNA constructs for injection into the germ line, and heritable gene changes occur in approximately 5% of progeny (Wood et al., 2011). While only a few studies have used ZFNs and TALENs to generate targeted deletions, these new protocol adjustments greatly improve efficiency. Help designing chimeric proteins can be found at http://www.zincfingers.org/ for ZFNs and at tale-nt.cac.cornell.edu for TALENs.
ZFN and TALEN protocol (Wood et al., 2011):
ZFN or TALEN mRNAs that target a gene of interest are injected at low pressure directly into the hermaphrodite gonad (Wood et al., 2011). The mRNAs have 5’ and 3’ UTRs to increase germ line translation efficacy, as well as a 5’ cap and a 3’ polyA tail. ZFN mRNAs are injected at 250 ng/µl per construct (total concentration of 500 ng/µl). TALENs are injected at 1500 ng/µl per construct. F1 animals with the highest likelihood of changes in a target gene arise 10-14 h after injection, with 3.5%-5% of the progeny acquiring a small indel during this 4 h period. F2 animals derived from these F1 animals are screened either by phenotype or by PCR (Wood et al., 2011).
CRISPR Protocol (Dickinson et al., 2013; Friedland et al., 2013):
CRISPR/Cas9 has been successfully adapted for use in C. elegans and allows for rapid mutant recovery, creation of tagged versions of endogenous loci, and the implementation of conditional knock-outs and knock-ins. Generation of the sgRNA requires minimal cloning (Friedland et al., 2013), and transgene assembly is facilitated by Gibson cloning techniques (Gibson et al., 2009). While some constraints on the target DNA may exist beyond the PAM requirement, CRISPR/Cas allows virtually any gene to be removed and edited with precision and fidelity.
The codon-optimized nuclear-localized (NLS) Cas9 protein and sgRNA may be introduced into the germline using a variety of methods, including as a DNA plasmid under the eft-3 and U6 promotors, respectively (Chen et al., 2013; Dickinson et al., 2013; Friedland et al., 2013; Tzur et al., 2013), as in vitro transcribed RNA (Chiu et al., 2013; Katic and Grosshans, 2013; Lo et al., 2013), a combination of both (Waaijers et al., 2013) or as a recombinant protein (Cho et al., 2013). Mutants can be isolated by phenotype screening or by PCR-based methods described previously. Additionally, a specific lesion or change may be inserted by HDR (Dickinson et al., 2013). For example, one elegant study introduced 4 T/A changes in the lin-31 locus to inactivate four MAPK phosphorylation sites and alter vulva development (Dickinson et al., 2013). For a review of the current variations, see (Frokjaer-Jensen, 2013).
For generation of mutant alleles of a gene of interest, clone a target DNA sequence containing G/A(N)19NGG, preferably within the coding region and towards the 5′ end of the gene (Friedland et al., 2013) under the U6 promoter. It is also useful if the region of interest to be deleted contains a restriction site for easier mutant screening (Friedland et al., 2013). Inject a DNA plasmid mixture into young adult hermaphrodites using standard techniques. This mixture should contain an NLS-Cas9 under the eft-3 promoter and a tbb-2 3′UTR for efficient germline expression (50 ng/μl), sgRNA sequence under the U6 promoter (45 ng/μl), and a co-injection marker (e.g., myo-3p::mCherry, 5 ng/μl) (Friedland et al., 2013). Isolate F1 animals expressing the co-injection marker and screen their progeny for deletions in the gene of interest using any of the previously discussed techniques.
A similar protocol is used for engineering in specific deletions, point mutations or GFP knock-ins off a transgene following a DSB (Dickinson et al., 2013). The repair transgene should contain at least 1.5 kb of homology regions flanking the desired change, and a selection marker, such as unc-119(+) or antibiotic selection, which may itself be flanked by loxP sites (Dickinson et al., 2013). A single plasmid containing coding sequences for Cas9 and the sgRNA is injected at 50 ng/μl. The repair template is injected at 10 ng/μl, and co-injection markers, including negative selection using peel-1, are included in the mix. Injected hermaphrodites are singled to individual plates and allowed to grow to starvation, in the case of unc-119(+) selection (Dickinson et al., 2013). Plates containing non-Unc progeny are heat shocked at 34oC for 4 h to induce peel-1 negative selection. Non-Unc animals are then singled to establish lines (Dickinson et al., 2013). While current studies in C. elegans have used a double-strand break to repair off a transgene, work in mice and human cell cultures suggest that using a nickase version or a double nickase system may increase the efficiency of transgene introduction (Ran et al., 2013). The Cas9 and sgRNA plasmids from (Friedland et al., 2013) and (Dickinson et al., 2013) are available on AddGene.
An array of random and directed genome alteration methods are available in C. elegans, and the choice of which method to use largely depends on the biological problem at hand. Chemical mutagenesis is generally useful for defining genes associated with a particular biological process, as are methods involving random generation of deletions and transposon insertions. Reverse genetic methods are utilized when the functions of specific genes are sought. Because all of the methods described here rely on random processes for mutation generation, screening of large numbers of animals is generally unavoidable. Thus, before embarking on mutagenesis, care should be taken to optimize screen design. Finally, it is imperative that the assignment of phenotype to gene be rigorously demonstrated. A good rule to follow is that two independent assays should corroborate an assignment. For example, rescue studies and allele identification, multiple alleles, or RNAi and allele isolation are all reasonable combinations confirming gene assignment.
We thank Don Moerman and Mark Edgley for sharing detailed EMS/ENU cocktail mutagenesis protocols. We thank Oliver Hobert for soliciting helpful comments on the manuscript. This work was funded in part by NIH grants NS081490, NS064273, and NS073121 to S.S., and also in part by NRSA Training Grant #GM066699 to L.M.K.
Ahringer, J. (2006). Reverse genetics. WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.47.1, http://www.wormbook.org. Article
Anderson, P. (1995). Mutagenesis. In Methods Cell Biology, H. F. Epstein and D. C. Shakes, ed. (San Diego, CA: Academic Press, Inc.), pp. 31-58. Abstract
Anderson, P., and Brenner, S. (1984). A selection for myosin heavy chain mutants in the nematode Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 81, 4470-4474. Abstract
Babcock, E.B., and Collins, J.L. (1929). Does natural ionizing radiation control rate of mutation? Proc. Natl. Acad. Sci. U. S. A. 15, 623-628. Abstract
Babu, P., and Brenner, S. (1981). Spectrum of 32P-induced mutants of Caenorhabditis elegans. Mutat. Res. 82, 269-273. Abstract
Bailly, V., Lamb, J., Sung, P., Prakash, S., and Prakash, L. (1994). Specific complex formation between yeast RAD6 and RAD18 proteins: a potential mechanism for targeting RAD6 ubiquitin-conjugating activity to DNA damage sites. Genes Dev. 8, 811-820. Abstract Article
Barrett, P.L., Fleming, J.T., and Gobel, V. (2004). Targeted gene alteration in Caenorhabditis elegans by gene conversion. Nat. Genet. 36, 1231-1237. Abstract
Barstead, R.J., and Moerman, D.G. (2006). C. elegans deletion mutant screening. In C. elegans: Methods and Applications, K. Strange, ed. (Totowa, NJ: Humana Press), pp. 51-58. Abstract
Bazopoulou, D., and Tavernarakis, N. (2009). The NemaGENETAG initiative: large scale transposon insertion gene-tagging in Caenorhabditis elegans. Genetica 137, 39-46. Abstract Article
Bessereau, J.L. (2006). Transposons in C. elegans. WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.70.1, http://www.wormbook.org. Article
Bessereau, J.L., Wright, A., Williams, D.C., Schuske, K., Davis, M.W., and Jorgensen, E.M. (2001). Mobilization of a Drosophila transposon in the Caenorhabditis elegans germ line. Nature 413, 70-74. Abstract Article
Beurdeley, M., Bietz, F., Li, J., Thomas, S., Stoddard, T., Juillerat, A., Zhang, F., Voytas, D.F., Duchateau, P., and Silva, G.H. (2013). Compact designer TALENs for efficient genome engineering. Nat. Commun. 4, 1762. Abstract Article
Boch, J., Scholze, H., Schornack, S., Landgraf, A., Hahn, S., Kay, S., Lahaye, T., Nickstadt, A., and Bonas, U. (2009). Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326, 1509-1512. Abstract Article
Boulin, T., and Bessereau, J.L. (2007). Mos1-mediated insertional mutagenesis in Caenorhabditis elegans. Nat. Protoc. 2, 1276-1287. Abstract
Brenner, S. (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71-94. Abstract
Brookes, P., and Lawley, P.D. (1961). The reaction of mono- and di-functional alkylating agents with nucleic acids. Biochem. J. 80, 496-503. Abstract
C. elegans Deletion Mutant Consortium. (2012). Large-scale screening for targeted knockouts in the Caenorhabditis elegans genome. G3 2, 1415-1425. Abstract Article
Cermak, T., Doyle, E.L., Christian, M., Wang, L., Zhang, Y., Schmidt, C., Baller, J.A., Somia, N.V., Bogdanove, A.J., and Voytas, D.F. (2011). Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 39, e82. Abstract Article
Chang, N., Sun, C., Gao, L., Zhu, D., Xu, X., Zhu, X., Xiong, J.W., and Xi, J.J. (2013). Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 23, 465-472. Abstract Article
Chen, C., Fenk, L.A., and de Bono, M. (2013). Efficient genome editing in Caenorhabditis elegans by CRISPR-targeted homologous recombination. Nucleic Acids Res. 41, e193. Abstract Article
Chiu, H., Schwartz, H.T., Antoshechkin, I., and Sternberg, P.W. (2013). Transgene-Free genome editing in Caenorhabditis elegans using CRISPR-Cas. Genetics 195, 1167-1171. Abstract Article
Cho, S.W., Kim, S., Kim, J.M., and Kim, J.S. (2013). Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 31, 230-232. Abstract Article
Cho, S.W., Lee, J., Carroll, D., Kim, J.S., and Lee, J. (2013). Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics 195, 1177-1180. Abstract Article
Chung, K., Crane, M.M., and Lu, H. (2008). Automated on-chip rapid microscopy, phenotyping and sorting of C. elegans. Nat. Methods 5, 637-643. Abstract Article
Colbert, T., Till, B.J., Tompa, R., Reynolds, S., Steine, M.N., Yeung, A.T., McCallum, C.M., Comai, L., and Henikoff, S. (2001). High-throughput screening for induced point mutations. Plant Physiol. 126, 480-484. Abstract Article
Cole, R.S. (1970). Light-induced cross-linking of DNA in the presence of a furocoumarin (psoralen). Studies with phage lambda, Escherichia coli, and mouse leukemia cells. Biochim. Biophys. Acta. 217, 30-39. Abstract Article
Collins, J., and Anderson, P. (1994). The Tc5 family of transposable elements in Caenorhabditis elegans. Genetics 137, 771-781. Abstract
Collins, J., Forbes, E., and Anderson, P. (1989). The Tc3 family of transposable genetic elements in Caenorhabditis elegans. Genetics 121, 47-55. Abstract
Collins, J., Saari, B., and Anderson, P. (1987). Activation of a transposable element in the germ line but not the soma of Caenorhabditis elegans. Nature 328, 726-728. Abstract
Cong, L., Ran, F.A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P.D., Wu, X., Jiang, W., Marraffini, L.A., and Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819-823. Abstract Article
Coulondre, C., and Miller, J.H. (1977). Genetic studies of the lac repressor. III. Additional correlation of mutational sites with specific amino acid residues. J. Mol. Biol. 117, 525-567. Abstract Article
Cradick, T.J., Fine, E.J., Antico, C.J., and Bao, G. (2013). CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 41, 9584-9592. Abstract Article
Crane, M.M., Stirman, J.N., Ou, C.Y., Kurshan, P.T., Rehg, J.M., Shen, K., and Lu, H. (2012). Autonomous screening of C. elegans identifies genes implicated in synaptogenesis. Nat. Methods 9, 977-980. Abstract Article
De Stasio, E.A., and Dorman, S. (2001). Optimization of ENU mutagenesis of Caenorhabditis elegans. Mutat. Res. 495, 81-88. Abstract Article
Denver, D.R., Dolan, P.C., Wilhelm, L.J., Sung, W., Lucas-Lledo, J.I., Howe, D.K., Lewis, S.C., Okamoto, K., Thomas, W.K., Lynch, M., and Baer, C.F. (2009). A genome-wide view of Caenorhabditis elegans base-substitution mutation processes. Proc. Natl. Acad. Sci. U. S. A. 106, 16310-16314. Abstract Article
Denver, D.R., Morris, K., Lynch, M., and Thomas, W.K. (2004). High mutation rate and predominance of insertions in the Caenorhabditis elegans nuclear genome. Nature 430, 679-682. Abstract Article
Dicarlo, J.E., Norville, J.E., Mali, P., Rios, X., Aach, J., and Church, G.M. (2013). Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 41, 4336-4343. Abstract Article
Dickinson, D.J., Ward, J.D., Reiner, D.J., and Goldstein, B. (2013). Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat. Methods 10, 1028-1034. Abstract Article
Doitsidou, M., Flames, N., Lee, A.C., Boyanov, A., and Hobert, O. (2008). Automated screening for mutants affecting dopaminergic-neuron specification in C. elegans. Nat. Methods 5, 869-872. Abstract Article
Doitsidou, M., Poole, R.J., Sarin, S., Bigelow, H., and Hobert, O. (2010). C. elegans mutant identification with a one-step whole-genome-sequencing and SNP mapping strategy. PLoS One 5, e15435. Abstract Article
Edgley, M., D'Souza, A., Moulder, G., McKay, S., Shen, B., Gilchrist, E., Moerman, D., and Barstead, R. (2002). Improved detection of small deletions in complex pools of DNA. Nucleic Acids Res. 30, e52. Abstract Article
Emmons, S.W., Yesner, L., Ruan, K.S., and Katzenberg, D. (1983). Evidence for a transposon in Caenorhabditis elegans. Cell 32, 55-65. Abstract Article
Fay, D.S. (2006). Classical genetic methods. WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.165.1, http://www.wormbook.org. Article
Fire, A., Xu, S., Montgomery, M.K., Kostas, S.A., Driver, S.E., and Mello, C.C. (1998). Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806-811. Abstract
Flibotte, S., Edgley, M.L., Chaudhry, I., Taylor, J., Neil, S.E., Rogula, A., Zapf, R., Hirst, M., Butterfield, Y., Jones, S.J., Marra, M.A., Barstead, R.J., and Moerman, D.G. (2010). Whole-genome profiling of mutagenesis in Caenorhabditis elegans. Genetics 185, 431-441. Abstract Article
Freeman, S.E., Hacham, H., Gange, R.W., Maytum, D.J., Sutherland, J.C., and Sutherland, B.M. (1989). Wavelength dependence of pyrimidine dimer formation in DNA of human skin irradiated in situ with ultraviolet light. Proc. Natl. Acad. Sci. U. S. A. 86, 5605-5609. Abstract
Friedland, A.E., Tzur, Y.B., Esvelt, K.M., Colaiacovo, M.P., Church, G.M. and Calarco, J.A. (2013). Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat. Methods 10, 741-743. Abstract Article
Frøkjaer-Jensen, C. (2013). Exciting prospects for precise engineering of Caenorhabditis elegans genomes with CRISPR/Cas9. Genetics 195, 635-642. Abstract Article
Frøkjaer-Jensen, C., Davis, M.W., Ailion, M., and Jorgensen, E.M. (2012). Improved Mos1-mediated transgenesis in C. elegans. Nat. Methods 9, 117-118. Abstract Article
Frøkjaer-Jensen, C., Davis, M.W., Hollopeter, G., Taylor, J., Harris, T.W., Nix, P., Lofgren, R., Prestgard-Duke, M., Bastiani, M., Moerman, D.G., and Jorgensen, E.M. (2010). Targeted gene deletions in C. elegans using transposon excision. Nat. Methods 7, 451-453. Abstract Article
Frøkjaer-Jensen, C., Davis, M.W., Hopkins, C.E., Newman, B.J., Thummel, J.M., Olesen, S.P., Grunnet, M., and Jorgensen, E.M. (2008). Single-copy insertion of transgenes in Caenorhabditis elegans. Nat. Genet. 40, 1375-1383. Abstract Article
Fu, Y., Foden, J.A., Khayter, C., Maeder, M.L., Reyon, D., Joung, J.K., and Sander, J.D. (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 31, 822-826. Abstract Article
Gengyo-Ando, K., and Mitani, S. (2000). Characterization of mutations induced by ethyl methanesulfonate, UV, and trimethylpsoralen in the nematode Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 269, 64-69. Abstract Article
Gibson, D.G., Young, L., Chuang, R.Y., Venter, J.C., Hutchison, C.A. 3rd, and Smith, H.O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 5, 343-345. Abstract Article
Gilchrist, E.J., O'Neil, N.J., Rose, A.M., Zetka, M.C., and Haughn, G.W. (2006). TILLING is an effective reverse genetics technique for Caenorhabditis elegans. BMC Genomics 7, 262. Abstract Article
Giordano-Santini, R., Milstein, S., Svrzikapa, N., Tu, D., Johnsen, R., Baillie, D., Vidal, M., and Dupuy, D. (2010). An antibiotic selection marker for nematode transgenesis. Nat. Methods 7, 721-723. Abstract Article
Goth, R., and Rajewsky, M.F. (1972). Ethylation of nucleic acids by ethylnitrosourea-1-14 C in the fetal and adult rat. Cancer Res. 32, 1501-1505. Abstract
Granger, L., Martin, E., and Segalat, L. (2004). Mos as a tool for genome-wide insertional mutagenesis in Caenorhabditis elegans: results of a pilot study. Nucleic Acids Res. 32, e117. Abstract
Greenwald, I.S., and Horvitz, H.R. (1980). unc-93(e1500): A behavioral mutant of Caenorhabditis elegans that defines a gene with a wild-type null phenotype. Genetics 96, 147-164. Abstract
Hartwell, L.H., Culotti, J., Pringle, J.R., and Reid, B.J. (1974). Genetic control of the cell division cycle in yeast. Science 183, 46-51. Abstract Article
Huang, C.G., Agre, P., Strange, K., and Lamitina, T. (2006). Isolation of C. elegans deletion mutants following ENU mutagenesis and thermostable restriction enzyme PCR screening. Mol. Biotech. 32, 83-86. Abstract
Hwang, W.Y., Fu, Y., Reyon, D., Maeder, M.L., Tsai, S.Q., Sander, J.D., Peterson, R.T., Yeh, J.R., and Joung, J.K. (2013). Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 31, 227-229. Abstract Article
Jacobson, J.W., Medhora, M.M., and Hartl, D.L. (1986). Molecular structure of a somatically unstable transposable element in Drosophila. Proc. Natl. Acad. Sci. U. S. A. 83, 8684-8688. Abstract Article
Jansen, G., Hazendonk, E., Thijssen, K.L., and Plasterk, R.H. (1997). Reverse genetics by chemical mutagenesis in Caenorhabditis elegans. Nat. Genet. 17, 119-121. Abstract Article
Jiang, W., Bikard, D., Cox, D., Zhang, F., and Marraffini, L.A. (2013). RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 31, 233-239. Abstract Article
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J.A., and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816-821. Abstract Article
Johnsen, R.C., and Baillie, D.L. (1988). Formaldehyde mutagenesis of the eT1 balanced region in Caenorhabditis elegans: dose-response curve and the analysis of mutational events. Mutat. Res. 201, 137-147. Abstract Article
Jorgensen, E.M., and Mango, S.E. (2002). The art and design of genetic screens: Caenorhabditis elegans. Nat. Rev. Genet. 3, 356-369. Abstract Article
Justice, M.J., Noveroske, J.K., Weber, J.S., Zheng, B., and Bradley, A. (1999). Mouse ENU mutagenesis. Hum. Mol. Genet. 8, 1955-1963. Abstract Article
Kagan, R.M., Niewmierzycka, A., and Clarke, S. (1997). Targeted gene disruption of the Caenorhabditis elegans L-isoaspartyl protein repair methyltransferase impairs survival of dauer stage nematodes. Arch. Biochem. Biophys. 348, 320-328. Abstract Article
Kamath, R.S., and Ahringer, J. (2003). Genome-wide RNAi screening in Caenorhabditis elegans. Methods 30, 313-321. Abstract Article
Katic, I., and Grosshans, H. (2013). Targeted heritable mutation and gene conversion by Cas9-CRISPR in Caenorhabditis elegans. Genetics 195, 1173-1176. Abstract Article
Ketting, R.F., Haverkamp, T.H., van Luenen, H.G., and Plasterk, R.H. (1999). mut-7 of C. elegans, required for transposon silencing and RNA interference, is a homolog of Werner syndrome helicase and RNaseD. Cell 99, 133-141. Abstract Article
Kim, Y.G., Cha, J., and Chandrasegaran, S. (1996). Hybrid restriction enzymes: zinc finger fusions to FokI cleavage domain. Proc. Natl. Acad. Sci. U. S. A. 93, 1156-1160. Abstract
Kruisselbrink, E., Guryev, V., Brouwer, K., Pontier, D.B., Cuppen, E., and Tijsterman, M. (2008). Mutagenic capacity of endogenous G4 DNA underlies genome instability in FANCJ-defective C. elegans. Curr. Biol. 18, 900-905. Abstract Article
Langmead, B., and Salzberg, S.L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357-359. Abstract Article
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S.L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. Abstract Article
Lawley, P.D., and Thatcher, C.J. (1970). Methylation of deoxyribonucleic acid in cultured mammalian cells by N-methyl-N'-nitro-N-nitrosoguanidine. Biochem. J. 116, 693-707. Abstract
Lesa, G.M. (2006). Isolation of Caenorhabditis elegans gene knockouts by PCR screening of chemically mutagenized libraries. Nat. Protoc. 1, 2231-2240. Abstract Article
Levitt, A., and Emmons, S.W. (1989). The Tc2 transposon in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 86, 3232-3236. Abstract
Liao, L.W., Rosenzweig, B., and Hirsh, D. (1983). Analysis of a transposable element in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 80, 3585-3589. Abstract
Lipinski, K.J., Farslow, J.C., Fitzpatrick, K.A., Lynch, M., Katju, V., and Bergthorsson, U. (2011). High spontaneous rate of gene duplication in Caenorhabditis elegans. Curr. Biol. 21, 306-310. Abstract Article
Liu, L.X., Spoerke, J.M., Mulligan, E.L., Chen, J., Reardon, B., Westlund, B., Sun, L., Abel, K., Armstrong, B., Hardiman, G., King, J., McCague, L., Basson, M., Clover, R., and Johnson, C.D. (1999). High-throughput isolation of Caenorhabditis elegans deletion mutants. Genome Res. 9, 859-867. Abstract Article
Lo, T.W., Pickle, C.S., Lin, S., Ralston, E.J., Gurling, M., Schartner, C.M., Bian, Q., Doudna, J.A., and Meyer, B.J. (2013). Precise and heritable genome editing in evolutionarily diverse nematodes using TALENs and CRISPR/Cas9 to engineer insertions and deletions. Genetics 195, 331-348. Abstract Article
Maduro, M., and Pilgrim, D. (1995). Identification and cloning of unc-119, a gene expressed in the Caenorhabditis elegans nervous system. Genetics 141, 977-988. Abstract
Mali, P., Yang, L., Esvelt, K.M., Aach, J., Guell, M., DiCarlo, J.E., Norville, J.E., and Church, G.M. (2013). RNA-guided human genome engineering via Cas9. Science 339, 823-826. Abstract Article
Martin, E., Laloux, H., Couette, G., Alvarez, T., Bessou, C., Hauser, O., Sookhareea, S., Labouesse, M., and Segalat, L. (2002). Identification of 1088 new transposon insertions of Caenorhabditis elegans: a pilot study toward large-scale screens. Genetics 162, 521-524. Abstract
Maydan, J.S., Flibotte, S., Edgley, M.L., Lau, J., Selzer, R.R., Richmond, T.A., Pofahl, N.J., Thomas, J.H., and Moerman, D.G. (2007). Efficient high-resolution deletion discovery in Caenorhabditis elegans by array comparative genomic hybridization. Genome Res. 17, 337-347. Abstract Article
Maydan, J.S., Okada, H.M., Flibotte, S., Edgley, M.L., and Moerman, D.G. (2009). De Novo identification of single nucleotide mutations in Caenorhabditis elegans using array comparative genomic hybridization. Genetics 181, 1673-1677. Abstract Article
McCallum, C.M., Comai, L., Greene, E.A., and Henikoff, S. (2000). Targeted screening for induced mutations. Nat. Biotechnol. 18, 455-457. Abstract Article
Meneely, P.M., and Herman, R.K. (1979). Lethals, steriles and deficiencies in a region of the X chromosome of Caenorhabditis elegans. Genetics 92, 99-115. Abstract
Miller, J.C., Tan, S., Qiao, G., Barlow, K.A., Wang, J., Xia, D.F., Meng, X., Paschon, D.E., Leung, E., Hinkley, S.J., Dulay, G.P., Hua, K.L., Ankoudinova, I., Cost, G.J., Urnov, F.D., Zhang, H.S., Holmes, M.C., Zhang, L., Gregory, P.D., and Rebar, E.J. (2011). A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 29, 143-148. Abstract Article
Minevich, G., Park, D.S., Blankenberg, D., Poole, R.J., and Hobert, O. (2012). CloudMap: a cloud-based pipeline for analysis of mutant genome sequences. Genetics 192, 1249-1269. Abstract Article
Mitani, S. (2009). Nematode, an experimental animal in the national BioResource project. Exp. Anim. 58, 351-356. Abstract
Moerman, D.G., and Baillie, D.L. (1981). Formaldehyde mutagenesis in the nematode Caenorhabditis elegans. Mutat. Res. 80, 273-279. Abstract Article
Mori, I., Benian, G.M., Moerman, D.G., and Waterston, R.H. (1988). Transposable element Tc1 of Caenorhabditis elegans recognizes specific target sequences for integration. Proc. Natl. Acad. Sci. U. S. A. 85, 861-864. Abstract
Mori, I., Moerman, D.G., and Waterston, R.H. (1988). Analysis of a mutator activity necessary for germline transposition and excision of Tc1 transposable elements in Caenorhabditis elegans. Genetics 120, 397-407. Abstract
Morton, J., Davis, M.W., Jorgensen, E.M., and Carroll, D. (2006). Induction and repair of zinc-finger nuclease-targeted double-strand breaks in Caenorhabditis elegans somatic cells. Proc. Natl. Acad. Sci. U. S. A. 103, 16370-16375. Abstract Article
Moscou, M.J., and Bogdanove, A.J. (2009). A simple cipher governs DNA recognition by TAL effectors. Science 326, 1501. Abstract Article
Muller, H.J. (1932). Further studies on the nature and causes of gene mutations. In Proceedings of the Sixth International Congress of Genetics, D. F. Jones, ed. (Ithaca, NY: Brooklyn Botanic Garden), pp. 213-255.
Nei, M. (2007). The new mutation theory of phenotypic evolution. Proc. Natl. Acad. Sci. U. S. A. 104, 12235-12242. Abstract Article
Ohnishi, K., and Ohnishi, T. (2004). The biological effects of space radiation during long stays in space. Biol. Sci. Space 18, 201-205. Abstract
Plasterk, R.H., and Groenen, J.T. (1992). Targeted alterations of the Caenorhabditis elegans genome by transgene instructed DNA double strand break repair following Tc1 excision. EMBO J. 11, 287-290. Abstract
Pontier, D.B., Kruisselbrink, E., Guryev, V., and Tijsterman, M. (2009). Isolation of deletion alleles by G4 DNA-induced mutagenesis. Nat. Methods 6, 655-657. Abstract Article
Praitis, V., Casey, E., Collar, D., and Austin, J. (2001). Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics 157, 1217-1226. Abstract
Ran, F.A., Hsu, P.D., Lin, C.Y., Gootenberg, J.S., Konermann, S., Trevino, A.E., Scott, D.A., Inoue, A., Matoba, S., Zhang, Y., and Zhang, F. (2013). Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380-1389. Abstract Article
Reedy, R.C., Arnold, J.R., and Lal, D. (1983). Cosmic-ray record in solar system matter. Science 219, 127-135. Abstract
Rezsohazy, R., van Luenen, H.G., Durbin, R.M., and Plasterk, R.H. (1997). Tc7, a Tc1-hitch hiking transposon in Caenorhabditis elegans. Nucleic Acids Res. 25, 4048-4054. Abstract Article
Robert, V., and Bessereau, J.L. (2007). Targeted engineering of the Caenorhabditis elegans genome following Mos1-triggered chromosomal breaks. EMBO J. 26, 170-183. Abstract Article
Robert, V.J. (2012). Engineering the Caenorhabditis elegans genome by Mos1-induced transgene-instructed gene conversion. In Mobile Genetic Elements: Protocols and Genomic Applications, Y. Bigot, ed. (Totowa, NJ: Humana Press), pp. 189-201. Abstract Article
Rosenbluth, R.E., Cuddeford, C., and Baillie, D.L. (1985). Mutagenesis in Caenorhabditis elegans. II. A spectrum of mutational events induced with 1500 r of gamma-radiation. Genetics 109, 493-511. Abstract
Rushforth, A.M., and Anderson, P. (1996). Splicing removes the Caenorhabditis elegans transposon Tc1 from most mutant pre-mRNAs. Mol. Cell Biol. 16, 422-429. Abstract
Sarin, S., Prabhu, S., O'Meara, M.M., Pe'er, I., and Hobert, O. (2008). Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nat. Methods 5, 865-867. Abstract Article
Semple, J.I., Biondini, L., and Lehner, B. (2012). Generating transgenic nematodes by bombardment and antibiotic selection. Nat. Methods 9, 118-119. Abstract Article
Semple, J.I., Garcia-Verdugo, R., and Lehner, B. (2010). Rapid selection of transgenic C. elegans using antibiotic resistance. Nat. Methods 7, 725-727. Abstract Article
Shaham, S. (2007). Counting mutagenized genomes and optimizing genetic screens in Caenorhabditis elegans. PLoS One 2, e1117. Abstract Article
Shaham, S. (2009). galign: a tool for rapid genome polymorphism discovery. PLoS One 4, e7188. Abstract Article
Sigurdson, D.C., Spanier, G.J., and Herman, R.K. (1984). Caenorhabditis elegans deficiency mapping. Genetics 108, 331-345. Abstract
Smith, H.E. (2011). Identifying insertion mutations by whole-genome sequencing. Biotechniques 50, 96-97. Abstract
Stewart, H.I., Rosenbluth, R.E., and Baillie, D.L. (1991). Most ultraviolet irradiation induced mutations in the nematode Caenorhabditis elegans are chromosomal rearrangements. Mutat. Res. 249, 37-54. Abstract Article
Tabara, H., Sarkissian, M., Kelly, W.G., Fleenor, J., Grishok, A., Timmons, L., Fire, A., and Mello, C.C. (1999). The rde-1 gene, RNA interference, and transposon silencing in C. elegans. Cell 99, 123-132. Abstract Article
Thompson, O., Edgley, M., Strasbourger, P., Flibotte, S., Ewing, B., Adair, R., Au, V., Chaudry, I., Fernando, L., Hutter, H., et al. (2013). The million mutation project: A new approach to genetics in Caenorhabditis elegans. Genome Res. 23, 1749-1762. Abstract Article
Tijsterman, M., Ketting, R.F., Okihara, K.L., Sijen, T., and Plasterk, R.H. (2002). RNA helicase MUT-14-dependent gene silencing triggered in C. elegans by short antisense RNAs. Science 295, 694-697. Abstract Article
Till, B.J., Burtner, C., Comai, L., and Henikoff, S. (2004). Mismatch cleavage by single-strand specific nucleases. Nucleic Acids Res. 32, 2632-2641. Abstract Article
Till, B.J., Reynolds, S.H., Greene, E.A., Codomo, C.A., Enns, L.C., Johnson, J.E., Burtner, C., Odden, A.R., Young, K., Taylor, N.E., et al. (2003). Large-scale discovery of induced point mutations with high-throughput TILLING. Genome Res. 13, 524-530. Abstract Article
Trent, C., Purnell, B., Gavinski, S., Hageman, J., Chamblin, C., and Wood, W.B. (1991). Sex-specific transcriptional regulation of the C. elegans sex-determining gene her-1. Mech. Dev. 34, 43-55. Abstract Article
Tzur, Y.B., Friedland, A.E., Nadarajan, S., Church, G.M., Calarco, J.A., and Colaiacovo, M.P. (2013). Heritable custom genomic modifications in Caenorhabditis elegans via a CRISPR-Cas9 System. Genetics 195, 1181-1185. Abstract Article
Vallin, E., Gallagher, J., Granger, L., Martin, E., Belougne, J., Maurizio, J., Duverger, Y., Scaglione, S., Borrel, C., Cortier, E., et al. (2012). A genome-wide collection of Mos1 transposon insertion mutants for the C. elegans research community. PLoS One 7, e30482. Abstract Article
van der Linden, A.M., and Plasterk, R.H.A. (2004). Shotgun cloning of transposon insertions in the genome of Caenorhabditis elegans. Comp. Funct. Genomics 5, 225-229. Abstract Article
Vastenhouw, N.L., Fischer, S.E., Robert, V.J., Thijssen, K.L., Fraser, A.G., Kamath, R.S., Ahringer, J., and Plasterk, R.H. (2003). A genome-wide screen identifies 27 genes involved in transposon silencing in C. elegans. Curr. Biol. 13, 1311-1316. Abstract Article
Vazquez-Manrique, R.P., Legg, J.C., Olofsson, B., Ly, S., and Baylis, H.A. (2010). Improved gene targeting in C. elegans using counter-selection and Flp-mediated marker excision. Genomics 95, 37-46. Abstract Article
Waaijers, S., Portegijs, V., Kerver, J., Lemmens, B.B., Tijsterman, M., van den Heuvel, S., and Boxem, M. (2013). CRISPR/Cas9-Targeted mutagenesis in Caenorhabditis elegans. Genetics 195, 1187-1191. Abstract Article
Wang, H., Yang, H., Shivalila, C.S., Dawlaty, M.M., Cheng, A.W., Zhang, F., and Jaenisch, R. (2013). One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910-918. Abstract Article
Wei, A., Yuan, A., Fawcett, G., Butler, A., Davis, T., Xu, S.Y., and Salkoff, L. (2002). Efficient isolation of targeted Caenorhabditis elegans deletion strains using highly thermostable restriction endonucleases and PCR. Nucleic Acids Res. 30, e110. Abstract Article
Wicks, S.R., Yeh, R.T., Gish, W.R., Waterston, R.H., and Plasterk, R.H. (2001). Rapid gene mapping in Caenorhabditis elegans using a high density polymorphism map. Nat. Genet. 28, 160-164. Abstract Article
Wiedenheft, B., Sternberg, S.H., and Doudna, J.A. (2012). RNA-guided genetic silencing systems in bacteria and archaea. Nature 482, 331-338. Abstract Article
Williams, D.C., Boulin, T., Ruaud, A.F., Jorgensen, E.M., and Bessereau, J.L. (2005). Characterization of Mos1-mediated mutagenesis in Caenorhabditis elegans: a method for the rapid identification of mutated genes. Genetics 169, 1779-1785. Abstract Article
Wood, A.J., Lo, T.W., Zeitler, B., Pickle, C.S., Ralston, E.J., Lee, A.H., Amora, R., Miller, J.C., Leung, E., Meng, X., et al. (2011). Targeted genome editing across species using ZFNs and TALENs. Science 333, 307. Abstract Article
Yandell, M.D., Edgar, L.G., and Wood, W.B. (1994). Trimethylpsoralen induces small deletion mutations in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 91, 1381-1385. Abstract Article
Yuan, J.Y., Finney, M., Tsung, N., and Horvitz, H.R. (1991). Tc4, a Caenorhabditis elegans transposable element with an unusual fold-back structure. Proc. Natl. Acad. Sci. U. S. A. 88, 3334-3338. Abstract Article
Zhao, Y., Lai, K., Cheung, I., Youds, J., Tarailo, M., Tarailo, S., and Rose, A. (2006). A mutational analysis of Caenorhabditis elegans in space. Mutat. Res. 601, 19-29. Abstract Article
Zuryn, S., Le Gras, S., Jamet, K., and Jarriault, S. (2010). A strategy for direct mapping and identification of mutations by whole-genome sequencing. Genetics 186, 427-430. Abstract Article
Zwaal, R.R., Broeks, A., van Meurs, J., Groenen, J.T., and Plasterk, R.H. (1993). Target-selected gene inactivation in Caenorhabditis elegans by using a frozen transposon insertion mutant bank. Proc. Natl. Acad. Sci. U. S. A. 90, 7431-7435. Abstract
*Edited by Oliver Hobert Last revised January 16, 2014, Published January 17, 2014. This chapter should be cited as: Kutscher L. M., Shaham S. Forward and reverse mutagenesis in C. elegans (January 17, 2014), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.167.1, http://www.wormbook.org.
Copyright: © 2014 Lena M. Kutscher and Shai Shaham. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: [email protected]
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.