Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
Despite the considerable advantages that C. elegans offers for studying gene function in vivo, this system is quite challenging for in vivo electrophysiological analysis of channel function, particularly in neurons. A major problem is that C. elegans neurons are confined in a pressurized and hard-to-penetrate cuticle. Recently, a method for culturing C. elegans embryonic cells has been developed and numerous researchers have already applied this option to study a variety of native ion channels and transporters using various configurations of the patch-clamp technique.
C. elegans embryonic cells are obtained from eggs harvested from synchronized gravid adults and then are dissociated using a combination of enzymatic treatment and manual pipetting. Once plated on a surface covered with peanut lectin, cells adhere and differentiate into neurons, muscle and epithelial cells. Cultured embryonic cells recapitulate the expression of differentiation markers and are found in the culture in proportion to their cell type in the mature embryo. Differentiated cells survive well for at least 2 weeks. It should be noted that postembryonic cells do not appear to be generated in these cultures. Cultures can be used for electrophysiological study, testing of pharmacological sensitivities, and for RNAi. C. elegans cell culture thus constitutes the basis for the application of experimental procedures that are not easily applicable to the intact nematode.
The value of C. elegans as a model system stems from the combination of two important facts: 1) basic cellular mechanisms are conserved between C. elegans and higher organisms, and 2) facile application of sophisticated molecular, genetic, and cell biological techniques can be executed against the backdrop of known cellular development, defined anatomy and a sequenced well-annotated genome. The nematode model, however, is not without its experimental challenges. C. elegans cells and organs are small when compared to their mammalian counterparts and they are enclosed within a pressurized and relatively impermeant cuticle. Together these anatomical constraints deterred execution of experimental approaches like single cell patch-clamping, construction of tissue-specific and cell-specific libraries, and drug evaluations for quite some time in C. elegans.
Elegant methods for patch-clamping of C. elegans muscle cells and neurons in situ have now been developed (Goodman et al., 1998; Richmond and Jorgensen, 1999). Unfortunately, these approaches are technically demanding and require substantial training and/or experience to execute. Sophisticated calcium imaging techniques have been successfully applied to live worms to provide insight into channel and transporter functions in vivo (Kerr et al., 2000; Suzuki et al., 2003; Hilliard et al., 2005), but these require specialized imaging equipment and software not broadly available in most labs. Moreover, manipulation of the extracellular environment and the administration of drugs through cuticle and gut barriers have remained challenges in live nematode analyses.
Several of these experimental limitations can be overcome using C. elegans cell culture. Christensen and colleagues recently reported a method for isolation and culture of C. elegans embryonic cells based on pioneering work of Laird Bloom, who established culture conditions for C. elegans cells during the preparation of his PhD thesis (Bloom, 1993; Christensen et al., 2002). Embryonic cells cultured with this method appear to differentiate and to recapitulate the properties that they express in vivo (Christensen et al., 2002; Estevez et al., 2003; Suzuki et al., 2003; Bianchi et al., 2004; Carvelli et al., 2004). Electrophysiology, calcium imaging and immunochemical techniques can be applied to cells isolated and cultured using this protocol (Christensen et al., 2002; Estevez et al., 2003; Suzuki et al., 2003; Bianchi et al., 2004; Carvelli et al., 2004; Frokjaer-Jensen et al., 2006), and a method for isolation of specific cells based on FACS sorting has been developed and used for the construction of at least two cell-specific cDNA libraries (Zhang et al., 2002; Fox et al., 2005; Touroutine et al., 2005). Moreover, protocols for gene knock-down by RNAi have been developed and successfully applied to cultured cells rendering this new method even more versatile (for more details on RNAi methods applied to embryonic cells see Methods in cell biology; Christensen et al., 2002; Park et al., 2005).
In conclusion the cell culture method adds a new dimension of versatility to the C. elegans model that can be exploited in the effort to bridge the gap between gene and function. In this chapter we review the history of method development, the protocol, and electrophysiological methods for analyses of cultured C. elegans cells.
Early attempts towards mass culture of C. elegans embryonic cells encountered difficulties such as low cell survival and loose attachment of the cells to substrates, which precluded the development of a reproducible protocol (Bloom, 1993). However, reports of successful small-scale culture of very young C. elegans embryonic cells were published as early as 1992. Goldstein and colleagues reported that blastomeres from embryos could be separated from other embryonic cells and later reassociated without affecting their ability to divide and differentiate normally (Goldstein, 1992). Edgar and colleagues developed a technique for in vitro culturing of embryos as young as the 1-cell stage that they used to study the early steps of development (Edgar, 1995). Embryonic cells were cultured by removing the eggshells using a combination of enzymatic digestion and manual dissociation. Cells that were prepared in this manner continued to divide, producing up to ~500 cells. Differentiation of the major lineages also occurred and neuronal processes were sometimes observed in the culture.
Leung and colleagues reported that when the intestinal precursor E blastomeres were cultured in vitro they could differentiate into epithelial cells that produced apical adherens junctions, expressed proteins in a polarized manner and surrounded an extracellular space to generate a structure resembling an intestinal lumen (Leung et al., 1999). Similarly, Buechner and colleagues described cells morphologically resembling canal cells following dissociation and culture of embryonic cells (Buechner et al., 1999). They also observed other cell types appearing to differentiate into neurons, muscles and epithelial cells.
This early work demonstrated the feasibility of culturing embryonic cells and constituted the basis for the thesis work of Laird Bloom aimed at defining optimal conditions for mass cell culture (Bloom, 1993). Ultimately, this work led to the cell culture protocol described by Christiansen and colleagues that is similar to that outlined below (Christensen et al., 2002).
C. elegans embryonic cells are prepared from eggs isolated from young gravid adults. Outlined here are protocols for preparation of large quantities of gravid adult worms, egg isolation and cell culture.
Worms are grown on 10 cm 8P plates (see recipe below) on a Na22 bacteria diet. Na22 bacteria, contrary to uracil-requiring OP50, grow in very thick layers and constitute an abundant food source for large quantities of worms.
Transfer worms to 8P plates from a starved plate. Do NOT chunk, instead wash worms off the starter plate with water and distribute the same amount of water + worms on each plate (500–1000 μl) to ensure plating the same number of worms/plate. Leave the plates uncovered for a few minutes until all water is absorbed and then store the plates at the appropriate temperature (usually 20°C).
8 P plates (1liter):
| NaCl | 3 g |
| Bactopeptone | 20 g |
| Agar | 25 g |
Autoclave for 30 min, let cool to 55–60°C and then add:
| Cholesterol (5mg/ml in EtOH) | 1 ml |
| 1 M CaCl2 | 1 ml |
| 1 M MgSO4 | 1 ml |
| KP buffer | 25 ml |
KP buffer stock (500ml):
| K2HPO4 | 5 g |
| KH2PO4 | 30 g |
| pH to 6.00 |
Pour into 10 cm Petri dishes (~ 30 ml/plate)
Next day, seed plates with Na22 bacteria grown overnight in 2XYT media (Sambrook, 1989). Instead of seeding only the center of the plate, seed the whole surface.
2XYT media (1 l):
| Tryptone | 16 g |
| Yeast extract | 10 g |
| NaCl | 5 g |
| Final pH 7.0 ± 0.2 at 25°C |
Typically one should begin with at least four 8P plates. Four confluent 8P plates should give enough worms for 12 wells of cultured cells (in 24-well plates). The goal is to get sterile eggs. This is achieved by multiple washes of the gravid adults first (in sterile water), bleaching of the adults to release eggs, and then washing of the eggs after isolation in sterile egg buffer.
Cells are plated on glass coverslips coated with peanut lectin. Before you start, prepare the coating of the glass coverslips with peanut lectin. Prepare a peanut lectin solution in sterile water at a concentration of 0.5 mg/ml and add it to sterile glass coverslips (12 mm diameter) placed at the bottom of the wells (200μl of solution is enough to cover the entire surface). Incubate until you are ready to plate the cells (20 min is enough). Right before you plate the cells, remove the peanut lectin solution and wash the wells once with sterile water. Remove the water completely before plating the cells. It is important to completely remove the lectin solution. Excess lectin on the coverslips causes cell clumping.
Wash adult worms off agar plates with Milli-Q water (for the first 2 washes it is not important that the water is sterile) into a 50 ml conical tube. Pellet worms by table-top centrifugation at 1200 rpm (~450g force) for 3 minutes.
Wash pelleted worms with Milli-Q water 3 or 4 more times (the last 1 or 2 times with sterile water) until the supernatant is clear of bacteria.
Transfer worms into a 15 ml conical tube and pellet them for the last time.
Remove the water and lyse the worm pellet with the following mixture of bleach and NaOH prepared immediately prior to use:
| Ingredient | Volume |
| Fresh Chlorox | 5.0 ml |
| 10 N NaOH | 1.25 ml |
| sterile H2O | 18.75 ml |
If you are starting from 6–8 8P plates use 5–6 ml of this mixture.
Rock worms gently during the bleach lysis. It is critical to monitor the extent of lysis by placing 1 drop of the mixture on a coverslip and examining worm bodies under the dissecting scope . The lysis reaction should be stopped when ~70% of the worms are broken at the middle. Typically, this takes about 5–10 minutes.
Stop the lysis reaction by filling the tube with egg buffer (118 mM NaCl, 48 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 25 mM Hepes, pH 7.3, 340 mOsm) and pellet the eggs and lysed worms by centrifugation at 1200 rpm for 3 minutes.
Remove the supernatant using a sterile plastic transfer pipette and wash 3 or 4 times with egg buffer. At the first wash the solution often appears yellow-- keep washing until the solution is clear. Also, make sure the pellet is completely re-suspended (you can vortex) in the egg buffer during each wash. Eggs tend to clump when in a pellet.
After the last centrifugation, carefully remove the buffer using a sterile plastic transfer pipette.
Eggs are separated from debris by centrifugation in a 30% sucrose solution. Resuspend the pelleted eggs and lysed worms in 2 ml of sterile egg buffer and then add 2 ml of a sterile 60% sucrose stock (in egg buffer). Mix this solution well (you can vortex).
Centrifuge the suspension at 1200 rpm (~450g) for 5–6 min.
Carefully remove tubes from the centrifuge. You should see eggs floating at the top of the solution. You should also see a small pellet consisting of worm carcasses. Using a P1000 pipettor and sterile tips, transfer the eggs into a sterile 15 ml conical tube. Sometimes many eggs can be also found a little below the meniscus, keep transferring the solution until you have transferred all the eggs.
Fill the tube with sterile egg buffer and re-pellet to remove the sucrose.
Wash pelleted eggs another 3 times with sterile egg buffer to completely remove sucrose.
From here on, all procedures are carried out in a laminar flow hood under sterile conditions.
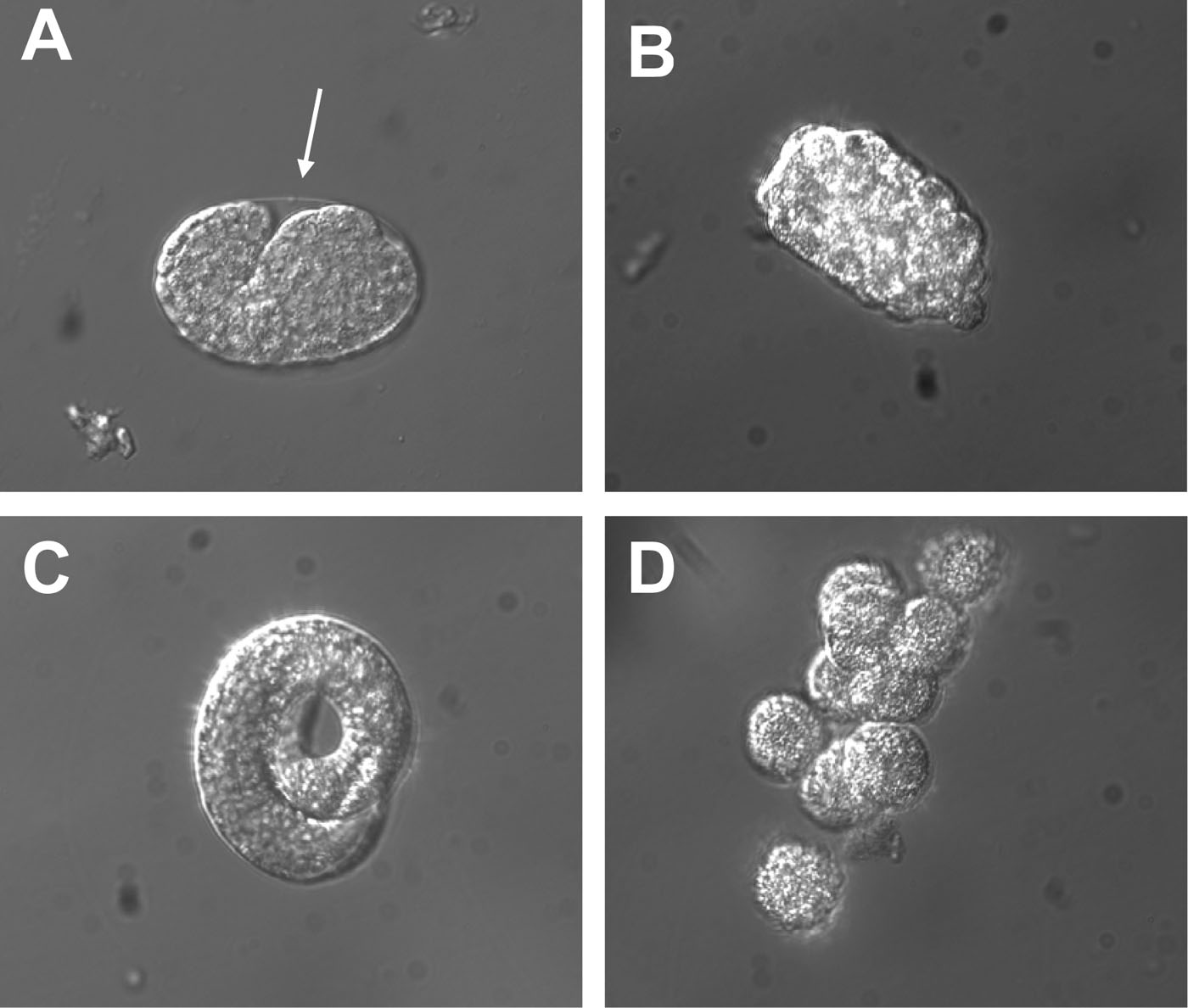
Resuspend pelleted eggs (Figure 1A) in 2 ml sterile egg buffer. Add 2 ml of 4 mg/ml chitinase (Sigma, C6137) and rock tubes for about 30 minutes at room temperature.
When approximately 80% of the eggshells are lysed, add 5 ml of complete L-15 cell culture medium to the 15 ml tube and transfer the eggs to a 5 cm diameter plate.
| Complete medium (500 ml) | |
| L-15 culture medium from Gibco | 500 ml |
| Fetal Bovine Serum (heat inactivated) from Gibco | 50 ml |
| Sucrose | 7.7 gr (45 mosm) |
| Pen/Strep (1:100) | 5 ml |
Pen/Strep stock solution (Gibco) contains 10,000 g/ml streptomycin and 10,000 units/ml of penicillin.
Filter using a 0.22 m pore filter after mixing.
Dissociate the cells using a syringe equipped with a needle. I usually use 27 gauge needles but other size needles are also fine. Aspirate the suspension and release it back into the plate a couple of times and then check under the microscope for the presence of dissociated cells (Figure 1D). Keep running the suspension through the needle until ~ 80% of the cells are dissociated. Do not aspirate air into the syringe, as it may damage the cell membrane.
|
Figure 1. Appearance of eggs prior and post chitinase treatment. (A) Photograph of an egg prior to chitinase treatment. The arrow points to the transparent eggshell that surrounds the embryo. (B) Photograph of an egg that has been treated with 2 mg/ml chitinase for 25 minutes. Note that the eggshell has been digested by the enzymatic treatment and that the embryo is now in direct contact with the egg buffer. (C) Photograph of a three-fold embryo released from the eggshell by chitinase treatment. (D) Embryonic cells dissociated from a chitinase-treated egg.
Filter the suspension through a sterile 5μm Durapore filter (Millipore). Run other 3–4 ml of L-15 media through the filter so that all the cells can be recovered from the filter.
Pellet the dissociated cell suspension by centrifugation at 1200 rpm for 15 minutes. Remove the supernatant by aspiration and resuspend the pellet in complete L-15 cell culture medium. The volume of media depends on the number of worms used to start the protocol and on the desired target cell density. For electrophysiological experiments it is useful to plate 12 wells of a 24 well plate, when starting with four confluent 8P plates (~230,000 cells/cm2). Use 1 ml of media/well. The cell density can be precisely determined using a haemocytometer.
Cells are grown on glass coverslips (12 mm diameter) covered with peanut lectin. Cells adhere well to polylysine also (Bloom, 1993; Christensen et al., 2002). Cells must adhere to the substrate in order to differentiate.
Morphological differentiation of cells and expression of GFP markers is usually complete within 24 h. Cell survive well for up to 3 weeks. We have determined expression of typical ionic currents in touch receptor neurons up to 19 days after plating, but we are not certain that cells maintain a fully differentiated state very late into the culture history. Each cell type should be carefully characterized for differentiation markers and cells are probably best used up to 1 week from preparation.
Early work on small scale embryo preps first revealed that that cultured embryonic cells behave like these cells in vivo in that they appeared to differentiate and expressed cell-specific GFP reporters (Goldstein, 1992; Bloom, 1993; Edgar, 1995; Buechner et al., 1999; Leung et al., 1999). Subsequent work by other laboratories extended these observations and supported this conclusion for at least some cells including some neurons, intestinal cells and muscle cells (Christensen et al., 2002; Estevez et al., 2003; Suzuki et al., 2003; Bianchi et al., 2004; Carvelli et al., 2004; Park et al., 2005). Other cell types remain uncharacterized and, although results are likely to be similar, investigators should verify that the cell type of interest behaves in culture as it does in vivo. Evidence that cultured cells behave in vitro as they do in vivo is summarized below.
Cultured cells undergo morphological differentiation. When embryonic cells are plated they are spherical in shape. Two to three hours after plating, some cells begin to send out processes. These are likely to be neuronal and muscle cells (White et al., 1986). Differentiation appears complete by 24 hours, although in some cases neuronal processes continue to elongate for another day or two (Bianchi et al., unpublished observations). Differentiation is strictly dependent on adhesion to a surface. Cells that fail to adhere tend to clump up and to maintain their spherical shape. It is not known whether clumped cells are able to survive for long periods of time in culture.
To the extent that it has been studied, culture cells assume the approximate morphology that they have in vivo. For example, in vivo C. elegans muscle cells have an unusual morphology in vivo--contrary to mammalian muscle cells that receive contact input from extended neuronal processes for formation of the synapses, C. elegans muscles actually send out processes that synapse onto the motor neurons (White et al., 1986). In vitro muscle cells somewhat recapitulate this branched-morphology, often assuming a Y-shaped structure (Christensen et al., 2002). Another example involves the gentle touch sensory neurons. In vivo embryonically-derived touch neurons (ALMs and PVMs) have only two neuronal processes with one being longer than the other. Cultured touch neurons (identified by expression of mec-4::gfp or mec-7::gfp, expressed exclusively or predominantly in these neurons, respectively) differentiate with this morphology (Figure 2A and B; Christensen et al., 2002; Bianchi et al., 2004). Finally, cultured canal cells and intestinal cells generate structures on the surface of the glass coverslip that resemble the lumen of the canal cell and of the intestine, respectively (Buechner et al., 1999; Leung et al., 1999; Bianchi and Driscoll, unpublished observations).
Cultured cells express cell-specific GFP markers. Morphological differentiation is accompanied by the activation of transcription of cell-specific genes. Indeed, this hypothesis is confirmed by the observation that cell-specific GFP reporters are expressed in culture and identify cells in proportion to their normal proportion within the embryo. For example, there are 81 muscle cells and 222 neurons in a L1 larva, of these 230 (76%) express unc-119::gfp. UNC-119 is a protein of unknown function expressed in all neurons and some muscle cells (Maduro and Pilgrim, 1995). When Christiansen and colleagues (Christensen et al., 2002) counted the number of unc-119::gfp-expressing cells in culture, they determined that they were 74–76% of all cells displaying a muscle or neuron-like morphology. Similarly, there are 6 gentle touch-sensing neurons that express the cell specific marker mec-4::gfp (Chalfie and Sulston, 1981). MEC-4 is a subunit of the mechanically gated Na+/Ca2+ channel that specifically transduces gentle touch (Driscoll and Chalfie, 1991; Bianchi and Driscoll, 2002; Bianchi et al., 2004; Suzuki et al., 2003; O'Hagan et al., 2005). Of these, 4 touch receptor neurons are embryonically-derived. Indeed in culture touch neurons constitute ~ 0.5 % of the total number of cells (4 of ~ 560 cells - 0.7%– in an intact 3 fold embryo are touch neurons) (Figure 2A; Suzuki et al., 2003; Bianchi et al., 2004).
Cultured cells express cell-specific markers. Embryonic cells in culture express proteins that they normally express in vivo. For example neuronal processes of cultured touch neurons stain with an antibody raised against acetylated-tubulin (Bianchi et al., 2004), a modification unique to the touch neurons (Fukushige et al., 1999). Thus, the cytoskeleton of the cultured touch neurons appears to be arranged and modified as in vivo (Figure 2B). Cultured touch neurons express also the toxic mutant channel MEC-4(d). mec-4(d) encodes a toxic hyperactive mec-4 mutant that causes death of touch neurons in vivo (Driscoll and Chalfie, 1991). Cultures prepared from a mec-4(d); pmec-4::GFP strain, fluorescent touch neurons appear initially but then degenerate. After 4 days of culture, while wild-type touch neurons are still alive and healthy, mec-4(d) touch neurons have completely disappeared from the culture (Figure 2C). Another important point is that dying touch neurons behave in culture as they do in vivo. mec-4(d)-induced necrosis can be rescued in vivo by maneuvers that either block the hyperactivated MEC-4 channel or that block release of Ca2+ from intracellular stores (Xu et al., 2001). The same results can be reproduced in vitro (Figure 2D; Bianchi et al., 2004). Taken together, these results indicate that mec-4(+ and d)-expressing neurons in culture differentiate and behave similarly to mec-4-expressing touch neurons in vivo (Bianchi et al., 2004).
|
Figure 2. Properties of cultured C. elegans touch neurons. (A) Primary culture of a C. elegans strain bzIs18 which expresses yellow cameleon 2.12 (yc2.12) in the mechanosensory neurons (Miyawaki et al., 1997; Kerr et al., 2000; Suzuki et al., 2003). All neurons expressing cameleon show neuronal morphology. Scale bar 10 micron. (B) Example of cultured touch neuron expressing GFP under the touch neuron specific promoter Pmec-4. On the bottom, the same neuron was stained with an antibody raised against acetylated-tubulin (Sigma); a unique modification to touch neurons in vivo (Fukushige et al., 1999). (C) C. elegans embryonic cells were cultured from Pmec-4::gfp and mec-4(d); Pmec-4::gfp nematodes. Cells were plated at the same density (~200,000 cells/cm2) in individual wells and the number of touch neurons/40x field (identified as expressing GFP) were counted 24 hours and 4 days after plating. Ten fields per sample were counted. (D) Experiment conducted similarly to panel C for WT and mec-4(d) touch neurons plus and minus genetic modification (calreticulin null mutant bz29) and pharmacological treatments (10μM dantrolene or 10μM amiloride). The number of touch neurons/40x field was determined 24 hours after plating. Ten fields per sample were counted. Modified from (Bianchi et al., 2004).
Christiansen and colleagues reported also that cultured muscle cells express muscle-specific proteins such as the myosin heavy chain UNC-54 and that neurons express neuronal markers such as synaptotagmin SNT-1 (Christensen et al., 2002). In addition ion channels detected in vivo by patch-clamp in a certain cell type can be observed in vitro as well (Christensen et al., 2002; O'Hagan et al., 2005; Park et al., 2005). For example in situ touch neurons express an outwardly rectifying voltage-dependent K+ current (O'Hagan et al., 2005) identical to the one that can be recorded in vitro under the same experimental conditions (Christensen et al., 2002; Suzuki et al., 2003) and cultured ASE neurons are endowed with a K+ current (Christensen et al., 2002; Park et al., 2005) (likely carried by a channel complex formed by K+ channel pore-subunit KVS-1 and accessory subunit MPS-1 (Bianchi et al., 2003; Park et al., 2005) identical to the current that is observed in vivo (Goodman et al., 1998; Pierce-Shimomura et al., 2001).
In conclusion cultured cell types examined in detail appear to differentiate, express specific proteins and modify these proteins in a cell-specific manner. Thus cultured embryonic cells constitute a reliable substrate for the application of techniques that benefit from having cells either easily accessible or isolated from all the rest of the organism, most notably electrophysiological techniques.
Electrophysiological techniques are based on the use of electrodes that need to be either in very close contact with the cell membrane or within the tissue. In the first instance the activity of channels within one single cell is recorded and in the second case electrical activity deriving from multiple cells can be measured. In either case accessibility is a major concern. Electrodes are made of glass and they have very fine and fragile tips which need to be free of obstructive material to maintain good conductive properties. Therefore access to the cell or tissue needs to be as free as possible of obstacles that may either damage or clog the electrodes. Worm cells are extremely inaccessible–neurons are often embedded in tissues and entry into the animal requires penetration of a thick, pressurized collagenous cuticle.
Elegant techniques have been developed for recording from neurons and muscle in situ. These techniques are based on three key steps: 1) immobilizing the worm by gluing it on a glass coverslip, 2) releasing the hydrostatic pressure by nicking the worm cuticle and 2) gaining access to the cells by opening up the cuticle or filleting the worm (Goodman et al., 1998; Richmond and Jorgensen, 1999). Each one of these steps is technically challenging and therefore time consuming.
Cultured cells can be studied employing electrophysiological techniques with much more ease primarily because they are accessible. Another advantage is that a large number of cells of any given type is present on a single coverslip and since plating density is controlled by the experimenter, this number can be adjusted as desired. However, culture cells are detached from the rest of the animal, have lost synaptic connections and are not guaranteed to recapitulate in vitro all their physiological properties. Thus, whenever any of these is a concern, cultured cells should not be employed and in situ electrophysiological should be preferred. Standard electrophysiological techniques such as cell-attached, inside-out, outside-out and whole-cell have been successfully applied to cultured cells and important information on worm channels and transporters have been gathered (Christensen and Strange, 2001; Christensen et al., 2002; Estevez et al., 2003; Suzuki et al., 2003; Carvelli et al., 2004 Park et al., 2005).
Detailed description of standard electrophysiological techniques and of the equipment needed to perform electrophysiological recordings can be found in many invaluable sources such as: 1) “Ion channels of excitable membranes” by Bertil Hille (Hille, 2001), 2) “Axon Guide: for electrophysiology and biophysics laboratory techniques” by Axon Instruments, Inc (free download at: http://www.axon.com/mr_Axon_Guide.html) and in many excellent reviews including several from Nobel Laureates E. Neher and B. Sakmann, the inventors of the patch-clamp technique (Neher and Sakmann, 1976; Neher et al., 1978; Sakmann and Neher, 1984; Neher and Sakmann, 1992). We will here only briefly review the various configurations of the patch-clamp technique and describe modifications that need to be applied to record channels from C. elegans cells.
Whole-cell configuration
In this configuration ionic currents at the level of the whole cell membrane are studied. A glass pipette with a tip diameter of a few micrometers (tip diameter depends on cell and current size, see (Hille, 2001)) is pressed on the cell membrane to form a seal of high electrical resistance (in the G range, a gigaseal). This allows gaining of electrical access to the cell. The disadvantage of this configuration is that the cellular content, containing factors and enzymes that may regulate ion channels, is lost right after membrane breakage.
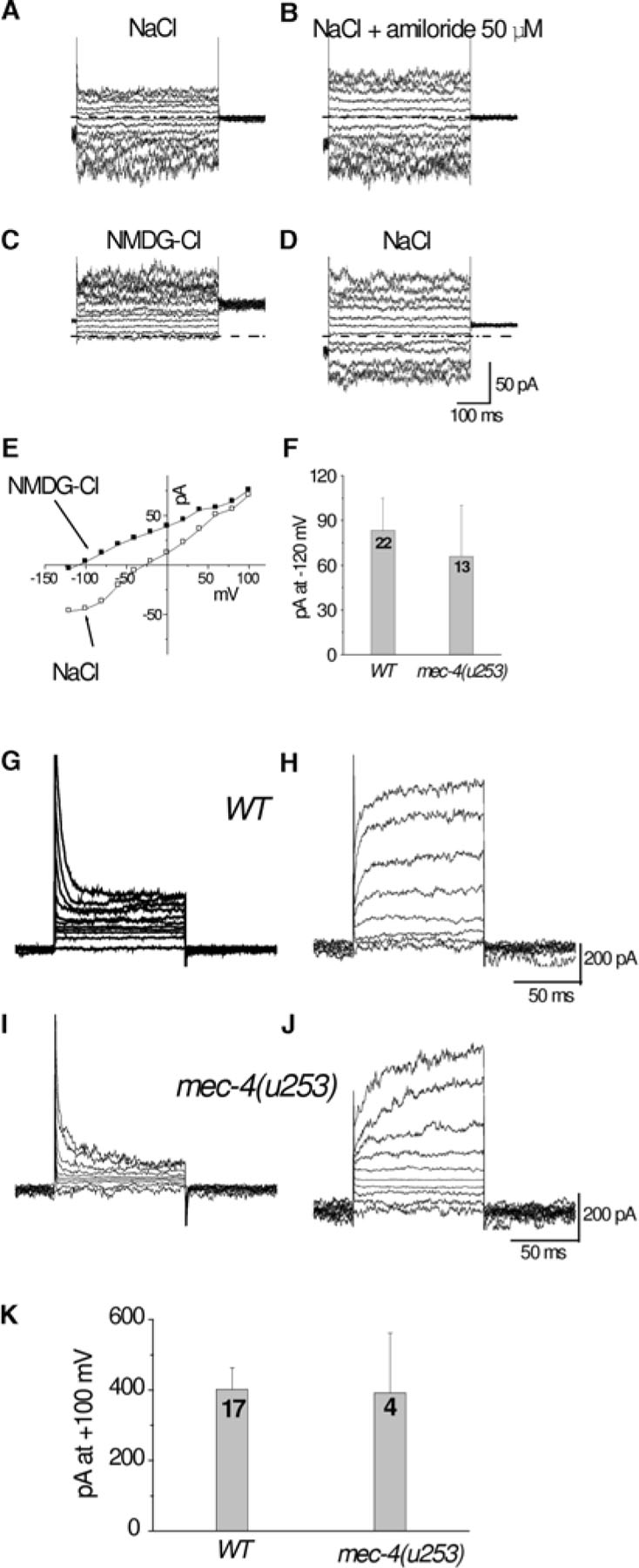
C. elegans cells are small (especially neurons) with a diameter of 1–2μm and therefore pipettes tip cannot be wider than 0.5μM. To maintain low series resistance (resistance caused partly by the size of the pipette opening) the shape of the pipette can be adjusted to have a small opening but a wide cone. Drs. Goodman and Lockery have developed a method for molding pipettes to this specific shape by pressure polishing (Goodman and Lockery, 2000). It should be noted though that if currents are small, like in most cases in C. elegans cells, series resistance will introduce only a very small error into the recordings (Figure 3A-D).
|
Figure 3. Whole-cell currents recorded from cultured C. elegans touch neurons. (A) Example of whole-cell currents recorded from a WT touch neuron perfused with a standard physiological saline. Voltage steps were from −120 to + 100 mV from a holding potential of −50 mV. (B) Currents elicited in the same neuron by the same voltage protocol as in A after application of 50μM amiloride. Note that the current is not affected by application of amiloride, indicating the lack of activity of amiloride sensitive channels under these experimental conditions. (C) Perfusion of the same cell with a solution in which NaCl was substituted by NMDG-Cl. Notice the disappearance of the inward component of the current during perfusion with NMDG-Cl indicating that this current is carried mainly by Na+ ions. (D) Upon washout and reperfusion with NaCl, the inward component reappears. (E) Current-voltage relationships of currents shown in A and C. (F) Average current at −120mV recorded from WT and mec-4(null) (mec-4(u253)) touch neurons. The average inward current is similar in the two genetic backgrounds reinforcing that MEC-4 is not active under these experimental conditions. (G) and (H) Families of outward currents likely carried by K+ recorded from two WT touch neurons perfused with a physiological saline. Voltage steps were from −80mV to +100mV from a holding potential of −50mV. WT and mec-4(u253) touch neurons are endowed with K+ currents of similar amplitudes. In most cases the K+ current activated and inactivated rapidly (G) and in a few touch neurons it activated slowly. The nature of this difference is not known at this point. Source: (Suzuki et al., 2003).
In C. elegans cells, the whole-cell configuration can be more easily obtained by electrical zapping, a short and high voltage impulse applied to the patch of membrane under the tip of the pipette that destabilizes the membrane and causes its breakage. Half a ms long 1.3 V pulses and 60 s long 7.2 V pulses work well (Goodman et al., 1998; Suzuki et al., 2003). Suction on the other hand in most cases damages or kills the cell. Perforation of the membrane using antibiotics such as nystatine or amphotericin B, another method that allows electrical access without cell damage (Horn and Marty, 1988), has been attempted on C. elegans cells but with little success (Christensen et al., 2002; Bianchi unpublished observation). As these antibiotics are known to perforate membranes containing cholesterol most efficiently (Horn and Marty, 1988; Marty and Finkelstein, 1975), these results suggest that C. elegans membrane composition may differ from mammals and possibly could contain little cholesterol. Examples of ionic currents recorded from cultured touch neurons using the whole-cell configuration are shown in Figure 3.
On cell (also known as cell-attached) patch-clamp mode. Cell-attached recordings are easily obtained in C. elegans cells. In this mode the patch of membrane under the tip of the pipette is not removed and the ionic currents resulting from individual openings of ion channels can be seen as discrete current steps in the recording. Examples of single channel recordings from cultured touch neurons are shown in Figure 4.
|
Figure 4. Single channels recorded from cultured touch neurons. Wild type and mec-4(null) touch neurons express a non-selective voltage-independent mechanosensitive channel. (A) Application of negative (but not positive) pressure to a touch neuron patch of membrane in the cell-attached configuration induces the activation of channels. (B) With release of the pressure the channels transition to the close configuration. The mechanically-gated channel is present in ~ 30 % of the patches examined. (C) Single channel openings of the mechanically-gated channel at voltages from −60 to + 100 mV during application of negative pressure. (D) Single-channel current-voltage relationship estimating a conductance of ~ 100 pS for the mechanically-gated channel.
Inside-out patch-clamp mode. In this mode once the cell-attached configuration is obtained, retreating the pipette will excise a patch with the internal membrane surface facing the bath solution (and the external surface facing the pipette solution). With this method the composition of the solution bathing the intracellular side of the membrane can be controlled, allowing for analysis of channels permeation properties and modulation by ions and factors such as ATP.
Outside-out patch-clamp mode. This configuration can be achieved by pulling the pipette away from the cell after obtaining the whole-cell configuration. A patch of membrane is excised with the extracellular side of the membrane facing the bath solution (and intracellular side facing the pipette filling solution). This method is generally used to measure single channel currents or currents from few channels in a configuration that preserves the native channel orientation in the membrane. This configuration allows studying of the effect of drugs that bind to the extracellular side of the channel on channel activity. Carvelli and colleagues successfully employed this configuration to measure single channel currents of native C. elegans dopamine transporter in cultured dopaminergic neurons after perfusing the patch with dopamine (Carvelli et al., 2004).
When patch-clamping C. elegans cells it is important to keep in mind that they require an osmolarity of ~ 340–345 mOsm to stay healthy (Christensen et al., 2002). The osmolarity of standard electrophysiological solutions should be adjusted to this value by adding sucrose.
The ionic composition of the fluid surrounding C. elegans cells is not known. Saline solutions for electrophysiological experiments, both in situ and on cultured cells, were designed based on the solution that supports pharyngeal pumping and normal electropharyngeograms in dissected animals (Avery et al., 1995).
Basal extracellular saline (in mM)
| NaCl | 145 |
| K-gluconate | 125 |
| KCl | 5 |
| KCl | 18 |
| CaCl2 | 1 |
| CaCl2 | 0.7 |
| MgCl2 | 5 |
| MgCl2 | 2 |
| HEPES | 10 |
| K2EGTA | 10 |
| D-Glucose | 20 |
| HEPES | 10 |
| Sucrose | to 345 mosm |
It is also important that glass capillaries used for pulling recording pipettes are chosen with care. Soft glass capillaries such as PG10165-4 from World Precision Instrument (Sarasota, FL) work well and don't need to be fire-polished (a procedure during which the tip of the pipette is heated and polished by a hot metal filament). Some researchers prefer to silanize the glass capillaries with dimethyl-dichloro silane (Sigma) (Christensen et al., 2002), but this is not strictly necessary.
Cultured cells rarely physically contact each other and are isolated from their normal neighbors. While this dissociation makes this system extremely powerful because cells are readily accessible, it is a disadvantage when channel function depends on the interaction with other cells or with the extracellular matrix. For example synaptic transmission cannot be studied on cultured cells since natural cell-cell contacts are lost and certain types (but not all) (Christensen and Strange, 2001) and (Bianchi and Driscoll, unpublished observations) of mechanically-gated channels may not gate in culture (Suzuki et al., 2003; O'Hagan et al., 2005).
For example mechanosensitive DEG/ENaC Na+/Ca2+ channel MEC-4 cannot be activated by mechanical forces (suction and osmotic pressure) in cultured cells (Suzuki et al., 2003), but are readily activated in vivo in intact worms (O'Hagan et al., 2005). MEC-4 is part of a multimeric channel complex together with MEC-10, another DEG/ENaC subunit, the stomatin-like protein MEC-2 and the paraoxonase-like protein MEC-6, and it is tethered to intracellular and extracellular proteins that are thought to exert gating tension on the channel during mechanical stimulation. On the extracellular side collagen MEC-5 and extracellular matrix proteins MEC-1 (a 1999 aa long protein endowed with a Kunitz-type domain and two EGF domains) and MEC-9 (a 834 aa long protein which contains several Kunitz-type domains, EGF repeats and a glutamic acid-rich domain) are bound to the channel and possibly push the channel open when mechanical forces are applied to the cuticle. The absence of these extracellular components in culture may disable channel gating.
Another problem that can be encountered when using cultured cells is that certain cellular structures may not develop correctly in culture. This is relevant especially when studying ion channels that are localized in specific subcellular domains and whose function may depend on their localization (Colbert et al., 1997; Tobin et al., 2002). For example it has been reported that the correct development of the sensory process of AWC neurons strictly depends on their physical interaction with the glial-like sheath cell (Taulant Bacaj, Shai Shaham ECWM 2004). In animals in which the sheath cell has been ablated the AWC sensory terminal loses its typical wing-like structure. Other sensory neurons terminals may be dependent on glial-like cells interaction in the same manner. Finally, post-embryonically derived cells cannot be studied in culture simply because they are not present in embryos used for the cell culture.
Isolated and in vitro cultured embryonic C. elegans cells have all the disadvantages of a culture system—namely loss of cell-cell and cell-extracellular matrix interaction, that may limit the study of certain phenomena or certain types of proteins. Despite these limitations cultured cells are well-suited for the application of techniques that benefit from isolation of cells from the rest of the organism. Cell cultures are particularly amenable for studying membrane proteins using electrophysiological techniques because they are easily accessible.
Research support from NIH grants R21-NS049511 to L.B. and R01-NS37955 to M.D. is acknowledged.
Avery, L., Raizen, D., and Lockery, S.R. (1995). Electrophysiological methods. In C. elegans: Modern biological analysis of an organism, H.F. Epstein and D.C. Shakes, eds. (San Diego, CA: Academic Press), 251–268. Abstract
Bianchi, L., and Driscoll, M. (2002). Protons at the gate: DEG/ENaC ion channels help us feel and remember. Neuron 34, 337–340. Abstract Article
Bianchi, L., Gerstbrein, B., Frokjaer-Jensen, C., Royal, D.C., Mukherjee, G., Royal, M.A., Xue, J., Schafer, W.R., and Driscoll, M. (2004). The neurotoxic MEC-4(d) DEG/ENaC sodium channel conducts calcium: implications for necrosis initiation. Nat. Neurosci. 7, 1337–1344. Abstract Article
Bianchi, L., Kwok, S., Driscoll M. and Sesti, F. (2003). A potassium channel-MiRP complex controls neurosensory function in Caenorhabditis elegans. J. Biol. Chem. 278, 12415–12424. Abstract Article
Bloom, L. (1993). Genetic and molecular analysis of genes required for axon outgrowth in Caenorhabditis elegans. PhD Thesis, Massachusetts Institute of Technology, Cambridge, MA.
Buechner, M., Hall, D.H., Bhatt, H., and Hedgecock, E.M. (1999). Cystic canal mutants in Caenorhabditis elegans are defective in the apical membrane domain of the renal (excretory) cell. Dev. Biol. 214, 227–241. Abstract Article
Carvelli, L., McDonald, P.W., Blakely, R.D., and Defelice, L.J. (2004). Dopamine transporters depolarize neurons by a channel mechanism. Proc. Natl. Acad. Sci. U.S.A. 101, 16046–16051. Abstract Article
Chalfie, M., and Sulston, J. (1981). Developmental genetics of the mechanosensory neurons of Caenorhabditis elegans. Dev. Biol. 82, 358–370. Abstract Article
Christensen, M., Estevez, A., Yin, X., Fox, R., Morrison, R., McDonnell, M., Gleason, C., Miller, D.M., and Strange, K. (2002). A primary culture system for functional analysis of C. elegans neurons and muscle cells. Neuron 33, 503–514. Abstract
Christensen, M.T., and Strange, K. (2001). Developmental regulation of a novel outwardly rectifying mechanosensitive anion channel in C. elegans. J. Biol. Chem. 21, 21. Abstract Article
Colbert, H.A., Smith, T.L., and Bargmann, C.I. (1997). OSM-9, a novel protein with structural similarity to channels, is required for olfaction, mechanosensation, and olfactory adaptation in Caenorhabditis elegans. J. Neurosci. 17, 8259–8269. Abstract Article
Driscoll, M., and Chalfie, M. (1991). The mec-4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degeneration. Nature 349, 588–593. Abstract Article
Edgar, L.G. (1995). Blastomere culture and analysis. Methods Cell Biol. 48, 303–321. Abstract
Estevez, A.Y., Roberts, R.K., and Strange, K. (2003). Identification of Store-independent and Store-operated Ca2+ Conductances in Caenorhabditis elegans Intestinal Epithelial Cells. J. Gen. Physiol. 122, 207–223. Abstract Article
Fox, R.M., Von Stetina, S.E., Barlow, S.J., Shaffer, C., Olszewski, K.L., Moore, J.H., Dupuy, D., Vidal, M., and Miller, D.M., 3rd (2005). A gene expression fingerprint of C. elegans embryonic motor neurons. BMC Genomics 6, 42. Abstract Article
Frokjaer-Jensen, C., Kindt, K.S., Kerr, R.A., Suzuki, H., Melnik-Martinez, K., Gerstbreih, B., Driscol, M., and Schafer, W.R. (2006). Effects of voltage-gated calcium channel subunit genes on calcium influx in cultured C. elegans mechanosensory neurons. J. Neurobiol. 66, 1125–1139. Abstract Article
Fukushige, T., Siddiqui, Z.K., Chou, M., Culotti, J.G., Gogonea, C.B., Siddiqui, S.S., and Hamelin, M. (1999). MEC-12, an a-tubulin required for touch sensitivity in C. elegans. J. Cell. Sci. 112, 395–403. Abstract
Goldstein, B. (1992). Induction of gut in Caenorhabditis elegans embryos. Nature 357, 255–257. Abstract Article
Goodman, M.B., Hall, D.H., Avery, L., and Lockery, S.R. (1998). Active currents regulate sensitivity and dynamic range in C. elegans neurons. Neuron 20, 763–772. Abstract Article
Goodman, M.B., and Lockery, S.R. (2000). Pressure polishing: a method for re-shaping patch pipettes during fire polishing. J. Neurosci. Methods 100, 13–15. Abstract Article
Hille, B. (2001). Ion channels of excitable membranes, Third edn. (Sunderland, Ma: Sinauer Associates, Inc.).
Hilliard, M.A., Apicella, A.J., Kerr, R., Suzuki, H., Bazzicalupo, P., and Schafer, W.R. (2005). In vivo imaging of C. elegans ASH neurons: cellular response and adaptation to chemical repellents. EMBO J. 24, 63–72. Abstract Article
Horn, R., and Marty, A. (1988). Muscarinic activation of ionic currents measured by a new whole-cell recording method. J. Gen. Physiol. 92, 145–159. Abstract Article
Kerr, R., Lev-Ram, V., Baird, G., Vincent, P., Tsien, R.Y., and Schafer, W.R. (2000). Optical imaging of calcium transients in neurons and pharyngeal muscle of C. elegans. Neuron 26, 583–594. Abstract Article
Leung, B., Hermann, G.J., and Priess, J.R. (1999). Organogenesis of the Caenorhabditis elegans intestine. Dev. Biol. 216, 114–134. Abstract Article
Maduro, M., and Pilgrim, D. (1995). Identification and cloning of unc-119, a gene expressed in the Caenorhabditis elegans nervous system. Genetics 141, 977–988. Abstract
Marty, A., and Finkelstein, A. (1975). Pores formed in lipid bilayer membranes by nystatin, Differences in its one-sided and two-sided action. J. Gen. Physiol. 65, 515–526. Abstract Article
Miyawaki, A., Llopis, J., Heim, R., McCaffery, J.M., Adams, J.A., Ikura, M., and Tsien, R.Y. (1997). Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 388, 882–887. Abstract Article
Neher, E., and Sakmann, B. (1976). Single-channel currents recorded from membrane of denervated frog muscle fibres. Nature 260, 799–802. Abstract Article
Neher, E., Sakmann, B., and Steinbach, J.H. (1978). The extracellular patch clamp: a method for resolving currents through individual open channels in biological membranes. Pflugers Arch. 375, 219–228. Abstract Article
Neher, E., and Sakmann, B. (1992). The patch clamp technique. Sci. Am. 266, 44–51. Abstract
O'Hagan, R., Chalfie, M., and Goodman, M.B. (2005). The MEC-4 DEG/ENaC channel of Caenorhabditis elegans touch receptor neurons transduces mechanical signals. Nat. Neurosci. 8, 43–50. Abstract Article
Park, K.H., Hernandez, L., Cai, S.Q., Wang, Y., and Sesti, F. (2005). A family of K+ channel ancillary subunits regulate taste sensitivity in Caenorhabditis elegans. J. Biol. Chem. 280, 21893–21899. Abstract Article
Pierce-Shimomura, J.T., Faumont, S., Gaston, M.R., Pearson, B.J., and Lockery, S.R. (2001). The homeobox gene lim-6 is required for distinct chemosensory representations in C. elegans. Nature 410, 694–698. Abstract Article
Richmond, J.E., and Jorgensen, E.M. (1999). One GABA and two acetylcholine receptors function at the C. elegans neuromuscular junction. Nat. Neurosci. 2, 791–797. Abstract Article
Sakmann, B., and Neher, E. (1984). Patch clamp techniques for studying ionic channels in excitable membranes. Annu. Rev. Physiol. 46, 455–472. Abstract Article
Sambrook J., Fritsch, E.F., and Maniatis, T. (1989). Molecular cloning: a laboratory manual, Second edition (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press).
Suzuki, H., Kerr, R., Bianchi, L., Frokjar-Jensen, C., Slone, D., Xue, J., Gerstbrein, B., Driscoll, M., and Schafer, W.R. (2003). In vivo imaging of C. elegans mechanosensory neurons demonstrates a specific role for the MEC-4 channel in the process of gentle touch sensation. Neuron 39, 1005–1017. Abstract Article
Tobin, D.M., Madsen, D.M., Kahn-Kirby, A., Peckol, E.L., Moulder, G., Barstead, R., and Bargmann, C.I. (2002). Combinatorial expression of TRPV channel proteins defines their sensory functions and subcellular localization in C. elegans. Neuron 35, 307–318. Abstract Article
Touroutine, D., Fox, R.M., Von Stetina, S.E., Burdina, A., Miller, D.M., 3rd, and Richmond, J.E. (2005). acr-16 encodes an essential subunit of the levamisole-resistant nicotinic receptor at the Caenorhabditis elegans neuromuscular junction. J. Biol. Chem. 280, 27013–27021. Abstract Article
White, J.G., Southgate, E., Thomson, J.N., and Brenner, S. (1986). The structure of the nervous system of Caenorhabditis elegans. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 314, 1–340. Abstract
*Edited by William J. Schafer. WormMethods editor, Victor Ambros. Last revised August 3, 2006. Published September 30, 2006. This chapter should be cited as: Bianchi, L. and Driscoll, M. Culture of embryonic C. elegans cells for electrophysiological and pharmacological analyses (September 30, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.122.1, http://www.wormbook.org.
Copyright: © 2006 Laura Bianchi and Monica Driscoll. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. Phone: 732-445 7183 or 87, Fax: 732-445 7192. E-mail: [email protected]
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.